CAUSEL: An epigenome and genome editing pipeline for establishing function of non-coding GWAS variants
Supporting Files

-
Sep 23 2015
-
Details
-
Alternative Title:Nat Med
-
Personal Author:Spisak, Sandor ; Lawrenson, Kate ; Fu, Yanfang ; Csabai, Istvan ; Cottman, Rebecca T. ; Haiman, Christopher ; Han, Ying ; Seo, Ji-Heui ; Lenci, Romina ; Li, Qiyuan ; Tisza, Viktoria ; Szallasi, Zoltan ; Herbert, Zachery T. ; Chabot, Matthew ; Pomerantz, Mark ; Solymosi, Norbert ; Gayther, Simon ; Joung, J. Keith ; Freedman, Matthew L.
-
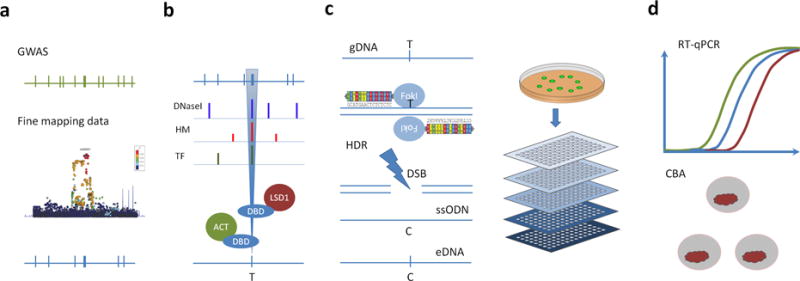
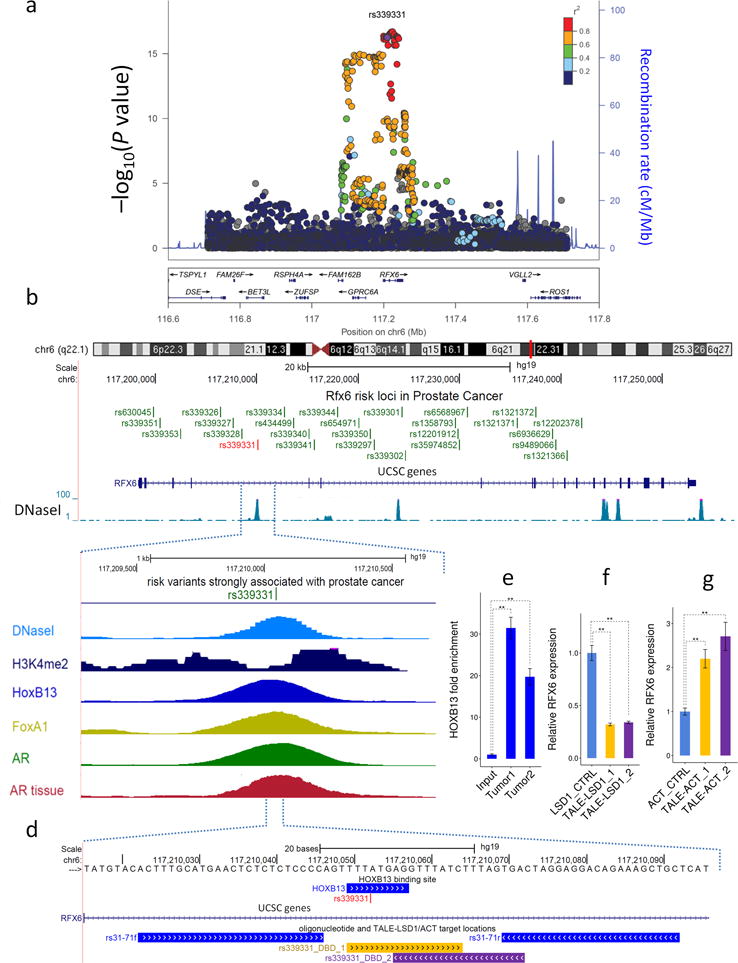
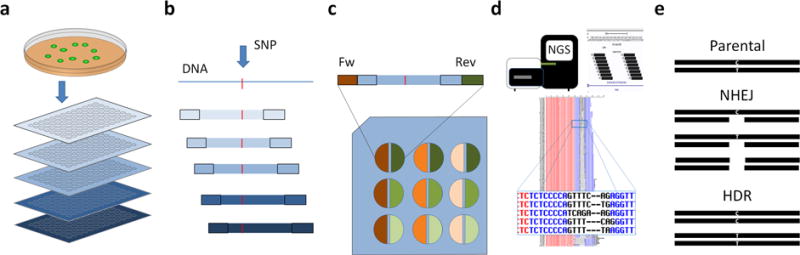
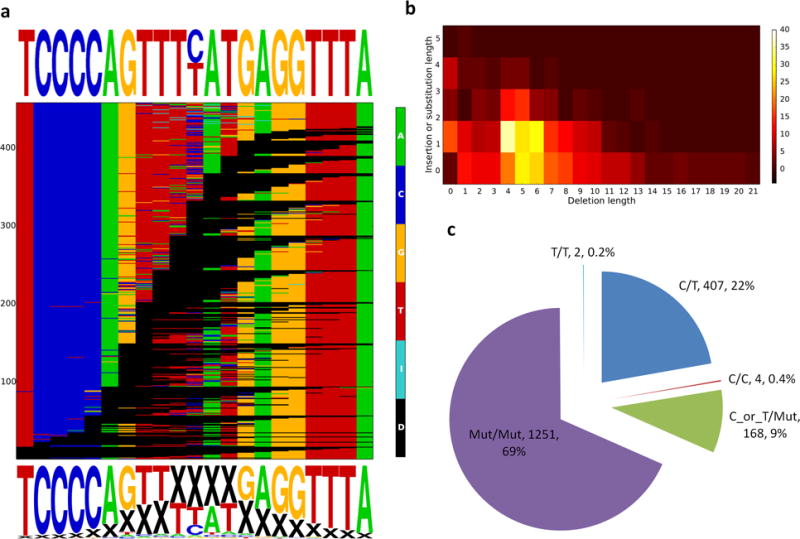
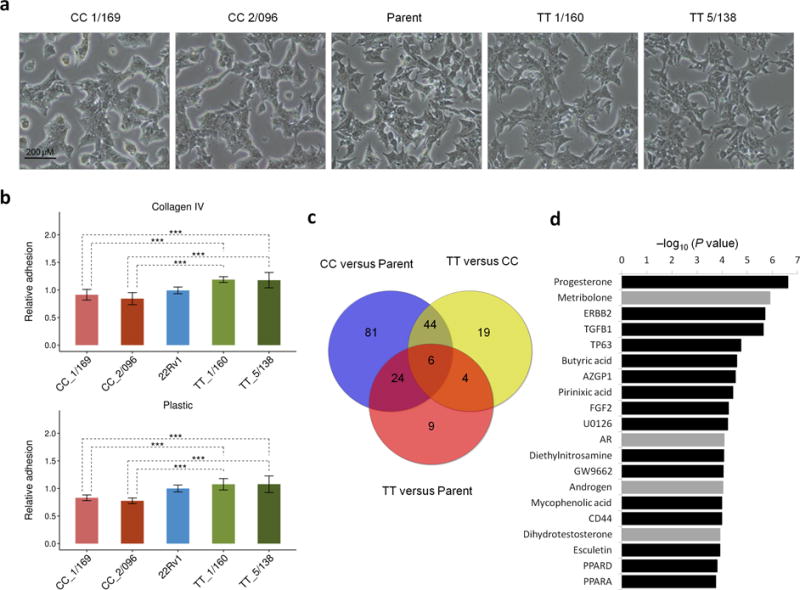
Description:The vast majority of disease-associated single-nucleotide polymorphisms (SNPs) mapped by genome-wide association studies (GWASs) are located in the non-protein-coding genome, but establishing the functional and mechanistic roles of these sequence variants has proven challenging. Here we describe a general pipeline in which candidate functional SNPs are first evaluated by fine mapping, epigenomic profiling, and epigenome editing, and then interrogated for causal function by using genome editing to create isogenic cell lines followed by phenotypic characterization. To validate this approach, we analyzed the 6q22.1 prostate cancer risk locus and identified rs339331 as the top-scoring SNP. Epigenome editing confirmed that the rs339331 region possessed regulatory potential. By using transcription activator-like effector nuclease (TALEN)-mediated genome editing, we created a panel of isogenic 22Rv1 prostate cancer cell lines representing all three genotypes (TT, TC, CC) at rs339331. Introduction of the 'T' risk allele increased transcription of the regulatory factor 6 (RFX6) gene, increased homeobox B13 (HOXB13) binding at the rs339331 region, and increased deposition of the enhancer-associated H3K4me2 histone mark at the rs339331 region compared to lines homozygous for the 'C' protective allele. The cell lines also differed in cellular morphology and adhesion, and pathway analysis of differentially expressed genes suggested an influence of androgens. In summary, we have developed and validated a widely accessible approach that can be used to establish functional causality for noncoding sequence variants identified by GWASs.
-
Subjects:
-
Source:Nat Med. 21(11):1357-1363.
-
Pubmed ID:26398868
-
Pubmed Central ID:PMC4746056
-
Document Type:
-
Funding:1K99CA184415-01/CA/NCI NIH HHS/United States ; DP1 GM105378/DP/NCCDPHP CDC HHS/United States ; DP1 GM105378/GM/NIGMS NIH HHS/United States ; DP1 OD006862/OD/NIH HHS/United States ; K08 DK002883/DK/NIDDK NIH HHS/United States ; P50 CA090381/CA/NCI NIH HHS/United States ; R01 CA129435/CA/NCI NIH HHS/United States ; R01 CA193910/CA/NCI NIH HHS/United States ; R01 GM107427/GM/NIGMS NIH HHS/United States ; R01 GM107427/GM/NIGMS NIH HHS/United States ; R01CA193910/CA/NCI NIH HHS/United States ; U19CA148112/CA/NCI NIH HHS/United States ; U19CA148537/CA/NCI NIH HHS/United States
-
Volume:21
-
Issue:11
-
Collection(s):
-
Main Document Checksum:urn:sha256:f6fd80a332773ea731516e17f6fa21e517db514fa2081eae654f014c91d01a90
-
Download URL:
-
File Type:
 [PDF
- 721.47 KB
]
[PDF
- 721.47 KB
]
Supporting Files

ON THIS PAGE

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
CDC STACKS serves as an archival repository of CDC-published products including
scientific findings,
journal articles, guidelines, recommendations, or other public health information authored or
co-authored by CDC or funded partners.
As a repository, CDC STACKS retains documents in their original published format to ensure public access to scientific information.
As a repository, CDC STACKS retains documents in their original published format to ensure public access to scientific information.
You May Also Like
COLLECTION
CDC Public Access