Comparative genomic analysis reveals occurrence of genetic recombination in virulent Cryptosporidium hominis subtypes and telomeric gene duplications in Cryptosporidium parvum
Supporting Files
Public Domain

-
Apr 18 2015
-
File Language:
English
Details
-
Alternative Title:BMC Genomics
-
Personal Author:
-
Description:Background
Cryptosporidium hominis is a dominant species for human cryptosporidiosis. Within the species, IbA10G2 is the most virulent subtype responsible for all C. hominis–associated outbreaks in Europe and Australia, and is a dominant outbreak subtype in the United States. In recent yearsIaA28R4 is becoming a major new subtype in the United States. In this study, we sequenced the genomes of two field specimens from each of the two subtypes and conducted a comparative genomic analysis of the obtained sequences with those from the only fully sequenced Cryptosporidium parvum genome.
Results
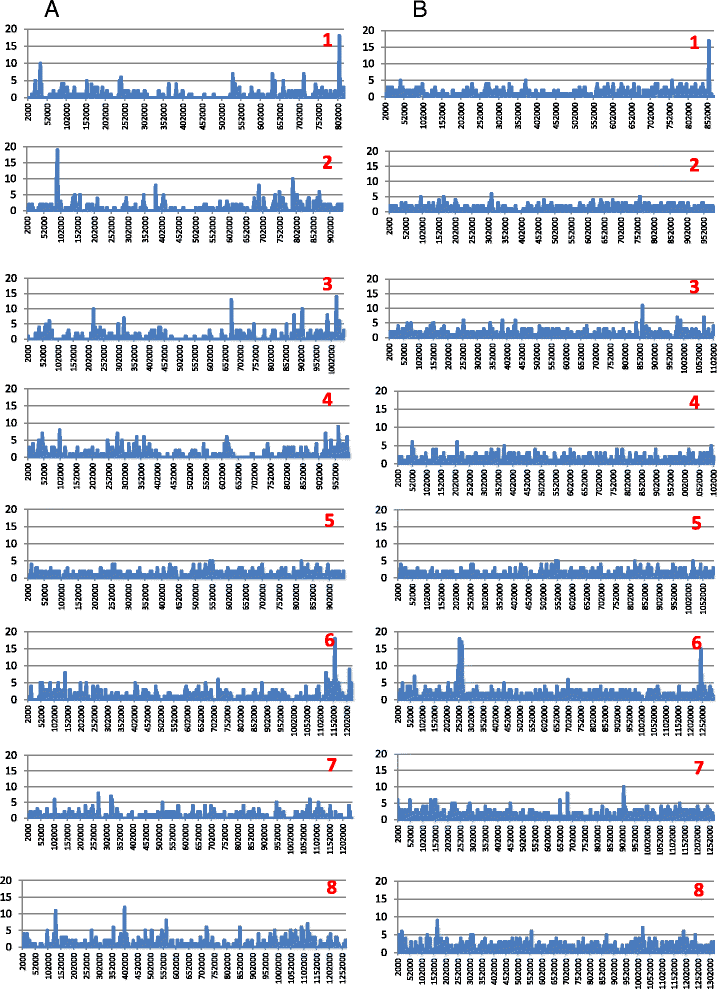
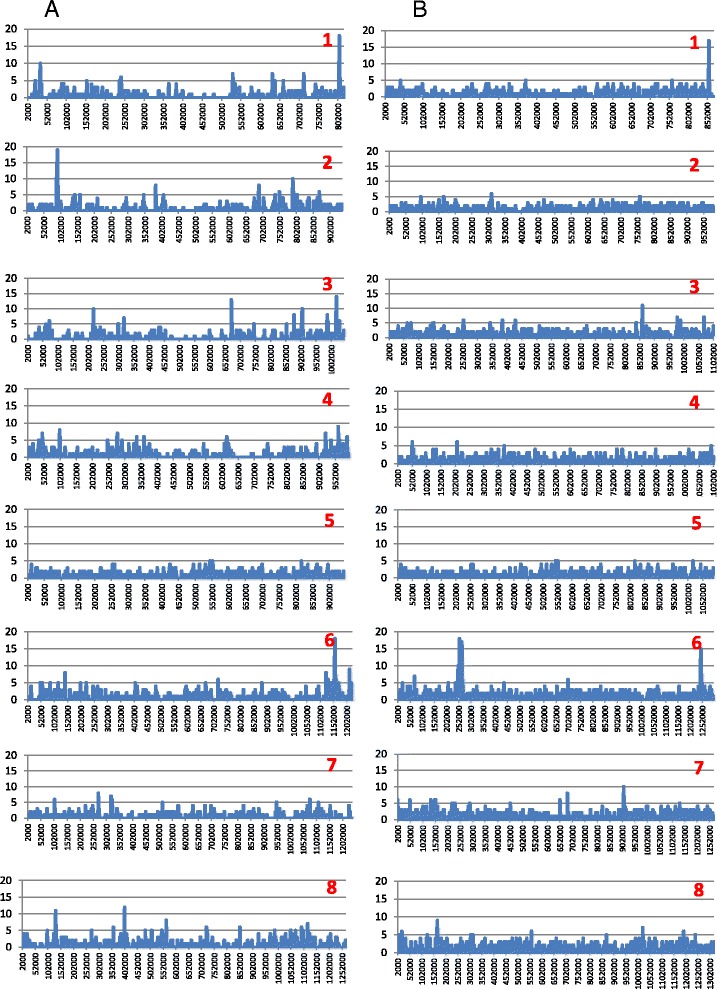
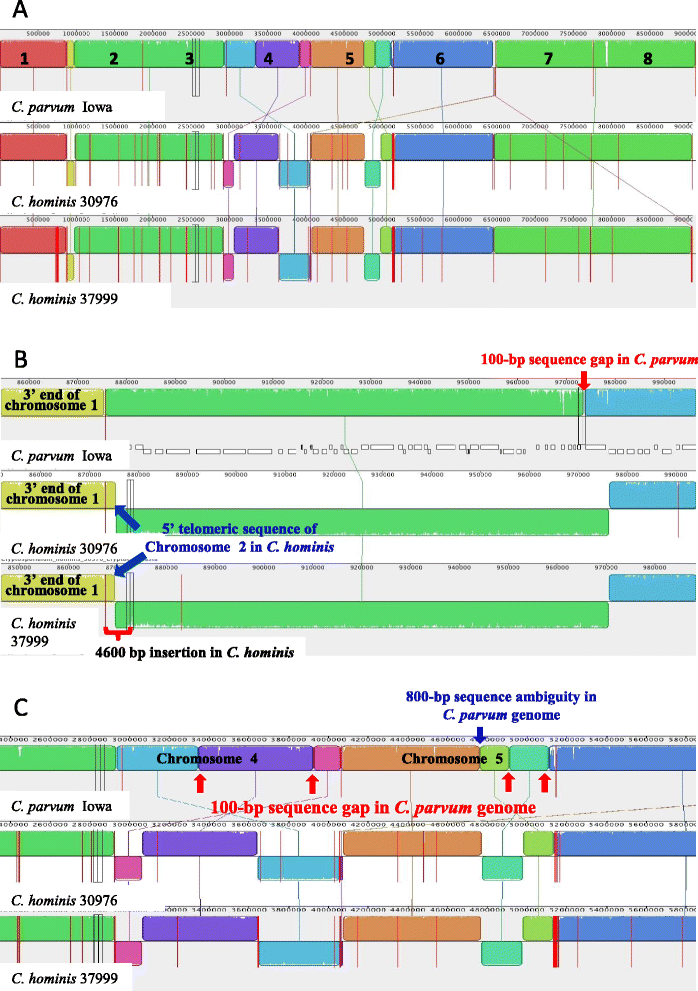
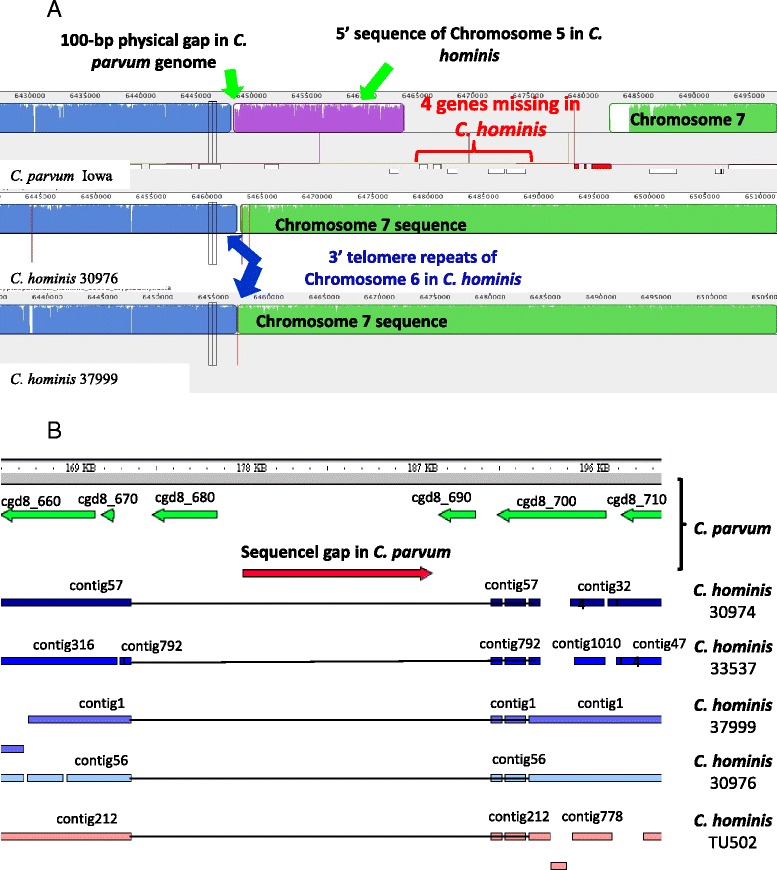
Altogether, 8.59-9.05 Mb of Cryptosporidium sequences in 45–767 assembled contigs were obtained from the four specimens, representing 94.36-99.47% coverage of the expected genome. These genomes had complete synteny in gene organization and 96.86-97.0% and 99.72-99.83% nucleotide sequence similarities to the published genomes of C. parvum and C. hominis, respectively. Several major insertions and deletions were seen between C. hominis and C. parvum genomes, involving mostly members of multicopy gene families near telomeres. The four C. hominis genomes were highly similar to each other and divergent from the reference IaA25R3 genome in some highly polymorphic regions. Major sequence differences among the four specimens sequenced in this study were in the 5′ and 3′ ends of chromosome 6 and the gp60 region, largely the result of genetic recombination.
Conclusions
The sequence similarity among specimens of the two dominant outbreak subtypes and genetic recombination in chromosome 6, especially around the putative virulence determinant gp60 region, suggest that genetic recombination plays a potential role in the emergence of hyper-transmissible C. hominis subtypes. The high sequence conservation between C. parvum and C. hominis genomes and significant differences in copy numbers of MEDLE family secreted proteins and insulinase-like proteases indicate that telomeric gene duplications could potentially contribute to host expansion in C. parvum.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-015-1517-1) contains supplementary material, which is available to authorized users.
-
Subjects:
-
Source:BMC Genomics. 16(1).
-
Document Type:
-
Volume:16
-
Issue:1
-
Collection(s):
-
Main Document Checksum:urn:sha256:85c27a5ff2bce6d0a9185d5c34f6888d6dc65d3659ab0103e344f72ab24dddb5
-
Download URL:
-
File Type:
 [PDF
- 2.93 MB
]
[PDF
- 2.93 MB
]
Supporting Files

File Language:
English
ON THIS PAGE

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
CDC STACKS serves as an archival repository of CDC-published products including
scientific findings,
journal articles, guidelines, recommendations, or other public health information authored or
co-authored by CDC or funded partners.
As a repository, CDC STACKS retains documents in their original published format to ensure public access to scientific information.
As a repository, CDC STACKS retains documents in their original published format to ensure public access to scientific information.
You May Also Like
COLLECTION
CDC Public Access