Next-generation sequencing reveals large connected networks of intra-host HCV variants
Supporting Files
Public Domain

-
Jul 14 2014
-
Details
-
Alternative Title:BMC Genomics
-
Personal Author:
-
Description:Background
Next-generation sequencing (NGS) allows for sampling numerous viral variants from infected patients. This provides a novel opportunity to represent and study the mutational landscape of Hepatitis C Virus (HCV) within a single host.
Results
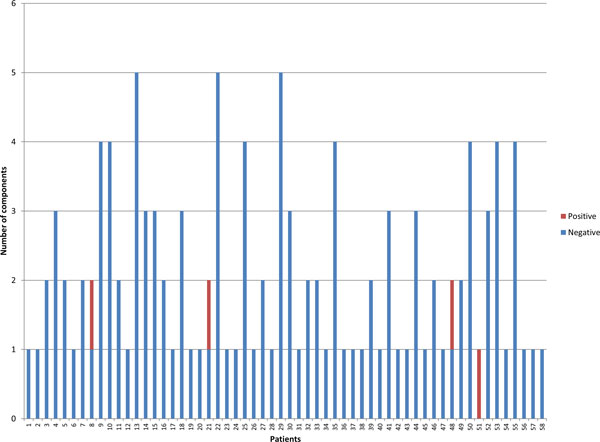
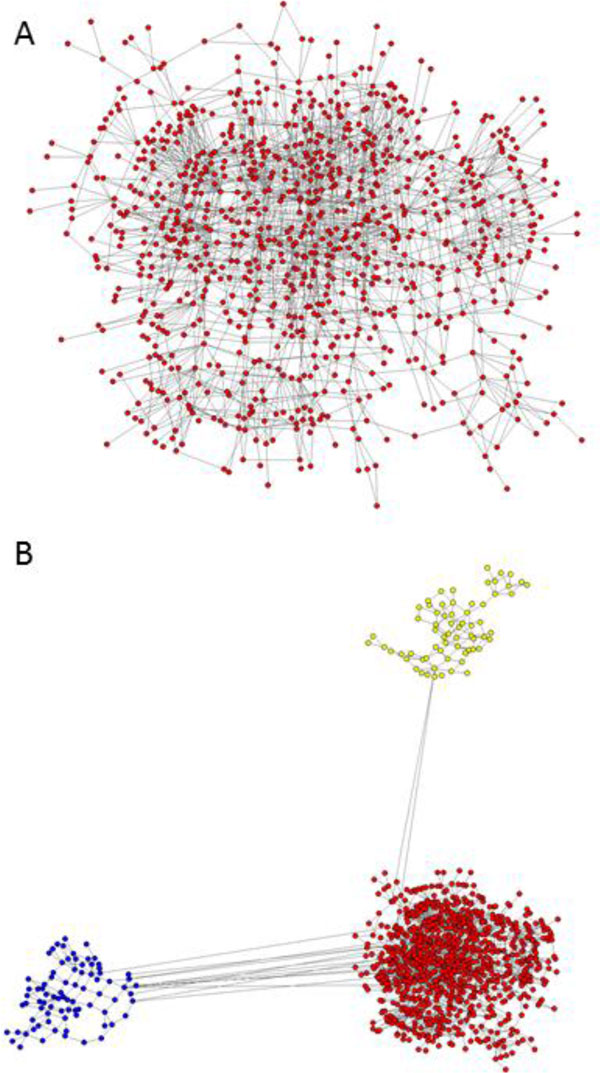
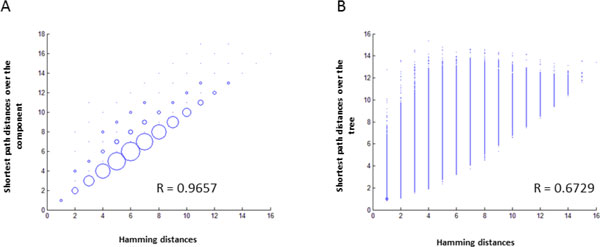
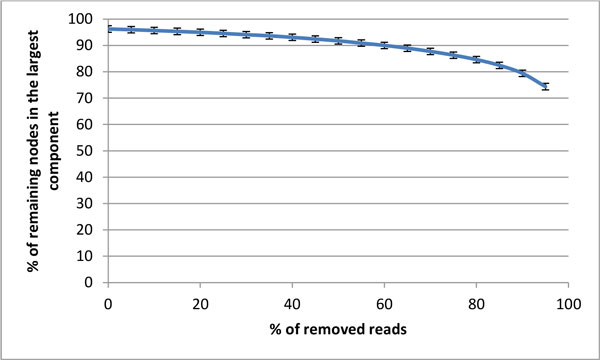
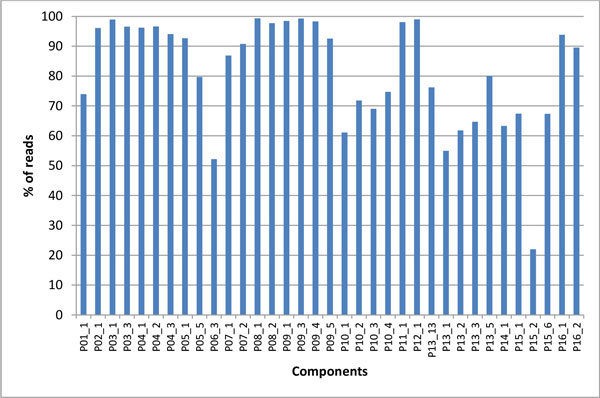
Intra-host variants of the HCV E1/E2 region were extensively sampled from 58 chronically infected patients. After NGS error correction, the average number of reads and variants obtained from each sample were 3202 and 464, respectively. The distance between each pair of variants was calculated and networks were created for each patient, where each node is a variant and two nodes are connected by a link if the nucleotide distance between them is 1. The work focused on large components having > 5% of all reads, which in average account for 93.7% of all reads found in a patient.
Conclusions
Most intra-host variants are organized into distinct single-mutation components that are: well separated from each other, represent genetic distances between viral variants, robust to sampling, reproducible and likely seeded during transmission events. Facilitated by NGS, large components offer a novel evolutionary framework for genetic analysis of intra-host viral populations and understanding transmission, immune escape and drug resistance.
-
Subjects:
-
Source:BMC Genomics. 2014; 15(Suppl 5):S4.
-
Document Type:
-
Volume:15
-
Collection(s):
-
Main Document Checksum:urn:sha256:fb2d918022983dbcda9437b9a7b9220ec670e897b663aaca7d76404c161848c8
-
Download URL:
-
File Type:
 [PDF
- 1.34 MB
]
[PDF
- 1.34 MB
]
Supporting Files

ON THIS PAGE

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
CDC STACKS serves as an archival repository of CDC-published products including
scientific findings,
journal articles, guidelines, recommendations, or other public health information authored or
co-authored by CDC or funded partners.
As a repository, CDC STACKS retains documents in their original published format to ensure public access to scientific information.
As a repository, CDC STACKS retains documents in their original published format to ensure public access to scientific information.
You May Also Like
COLLECTION
CDC Public Access