BMScan: using whole genome similarity to rapidly and accurately identify bacterial meningitis causing species
Supporting Files
Public Domain

-
8 15 2018
-
File Language:
English
Details
-
Alternative Title:BMC Infect Dis
-
Personal Author:
-
Description:Background
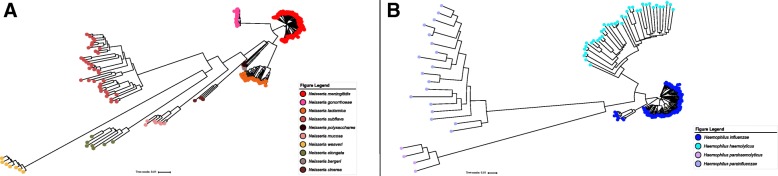
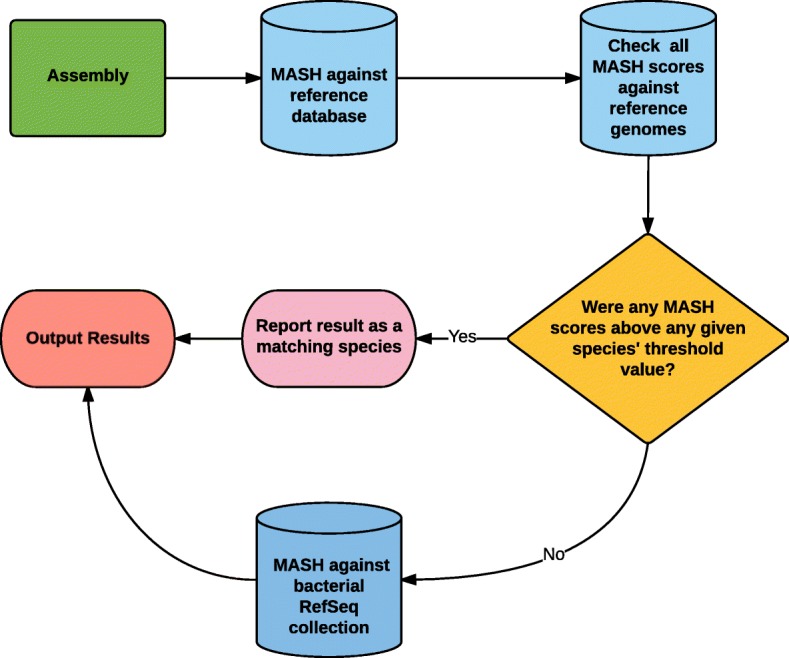
Bacterial meningitis is a life-threatening infection that remains a public health concern. Bacterial meningitis is commonly caused by the following species: Neisseria meningitidis, Streptococcus pneumoniae, Listeria monocytogenes, Haemophilus influenzae and Escherichia coli. Here, we describe BMScan (Bacterial Meningitis Scan), a whole-genome analysis tool for the species identification of bacterial meningitis-causing and closely-related pathogens, an essential step for case management and disease surveillance. BMScan relies on a reference collection that contains genomes for 17 focal species to scan against to identify a given species. We established this reference collection by supplementing publically available genomes from RefSeq with genomes from the isolate collections of the Centers for Disease Control Bacterial Meningitis Laboratory and the Minnesota Department of Health Public Health Laboratory, and then filtered them down to a representative set of genomes which capture the diversity for each species. Using this reference collection, we evaluated two genomic comparison algorithms, Mash and Average Nucleotide Identity, for their ability to accurately and rapidly identify our focal species.
Results
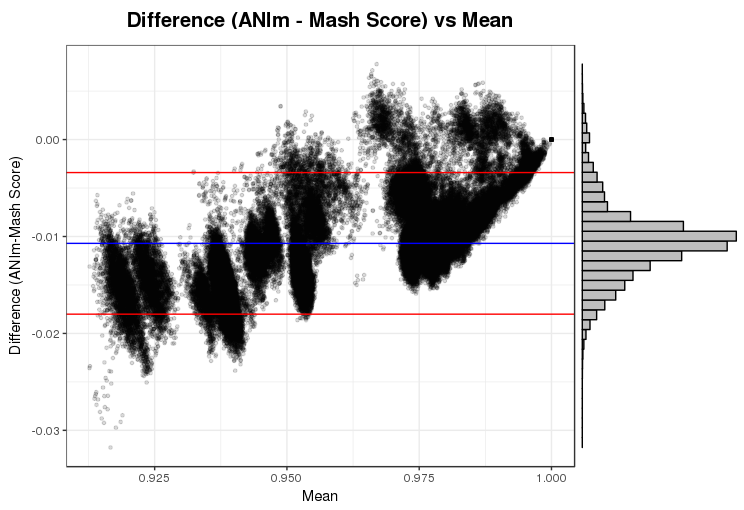
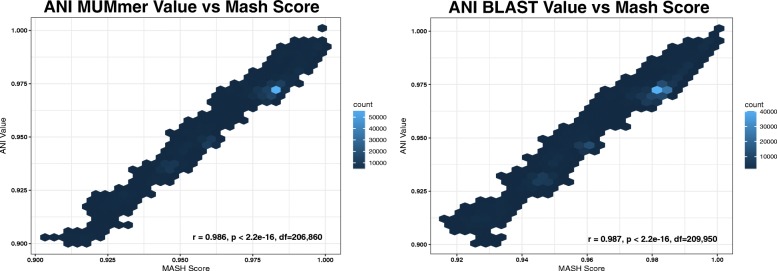
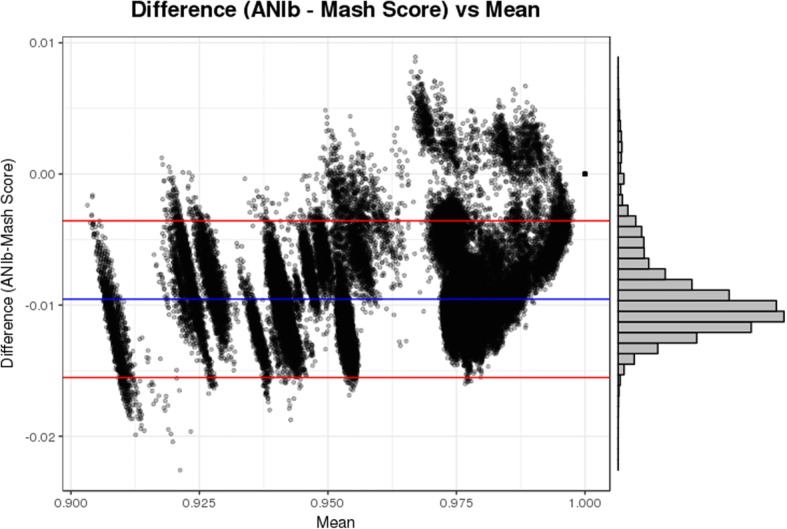
We found that the results of Mash were strongly correlated with the results of ANI for species identification, while providing a significant reduction in run-time. This drastic difference in run-time enabled the rapid scanning of large reference genome collections, which, when combined with species-specific threshold values, facilitated the development of BMScan. Using a validation set of 15,503 genomes of our species of interest, BMScan accurately identified 99.97% of the species within 16 min 47 s.
Conclusions
Identification of the bacterial meningitis pathogenic species is a critical step for case confirmation and further strain characterization. BMScan employs species-specific thresholds for previously-validated, genome-wide similarity statistics compiled from a curated reference genome collection to rapidly and accurately identify the species of uncharacterized bacterial meningitis pathogens and closely related pathogens. BMScan will facilitate the transition in public health laboratories from traditional phenotypic detection methods to whole genome sequencing based methods for species identification.
Electronic supplementary material
The online version of this article (10.1186/s12879-018-3324-1) contains supplementary material, which is available to authorized users.
-
Subjects:
-
Source:BMC Infect Dis. 18
-
Pubmed ID:30111301
-
Pubmed Central ID:PMC6094466
-
Document Type:
-
Volume:18
-
Collection(s):
-
Main Document Checksum:urn:sha-512:3eaf62fa21df46bdd05d034dc0145eb9d15bacd0fc2037ea06f2f577bd5158ee6471905178455045b032e50fcd919ec58c3eaa9f422fdb638846ce9a02a002d2
-
Download URL:
-
File Type:
 [PDF
- 1.16 MB
]
[PDF
- 1.16 MB
]
Supporting Files

File Language:
English
ON THIS PAGE

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
CDC STACKS serves as an archival repository of CDC-published products including
scientific findings,
journal articles, guidelines, recommendations, or other public health information authored or
co-authored by CDC or funded partners.
As a repository, CDC STACKS retains documents in their original published format to ensure public access to scientific information.
As a repository, CDC STACKS retains documents in their original published format to ensure public access to scientific information.
You May Also Like
COLLECTION
Stephen B. Thacker CDC Library