An Assessment of Different Genomic Approaches for Inferring Phylogeny of Listeria monocytogenes
Supporting Files
Public Domain

-
Nov 29 2017
Details
-
Alternative Title:Front Microbiol
-
Personal Author:
-
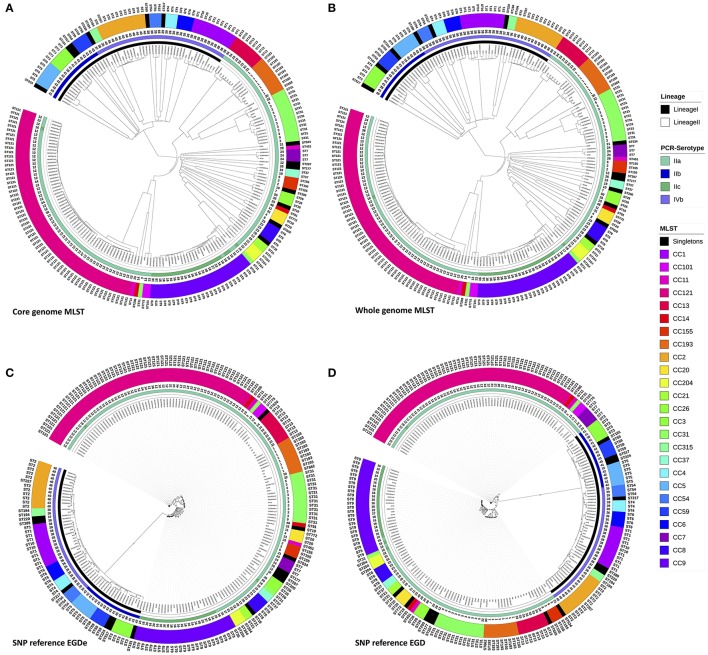
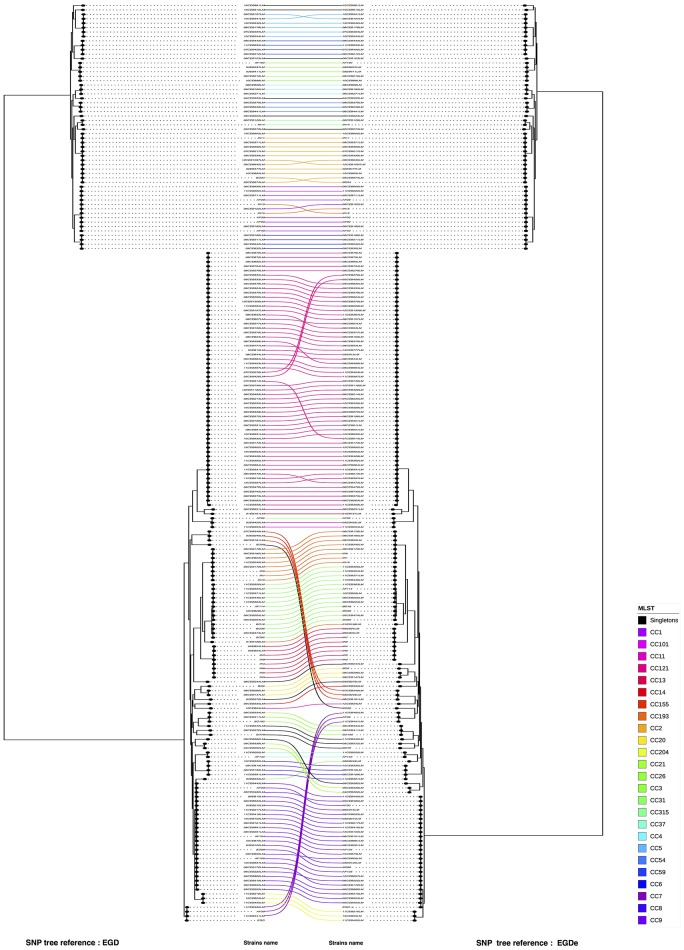
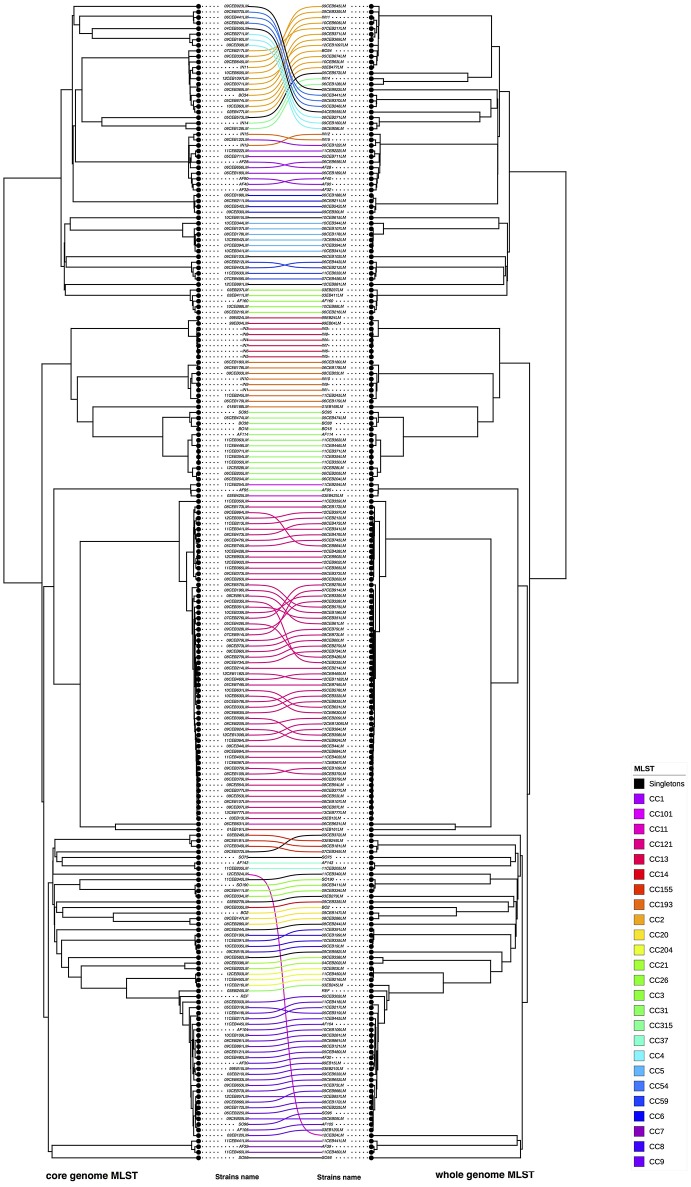
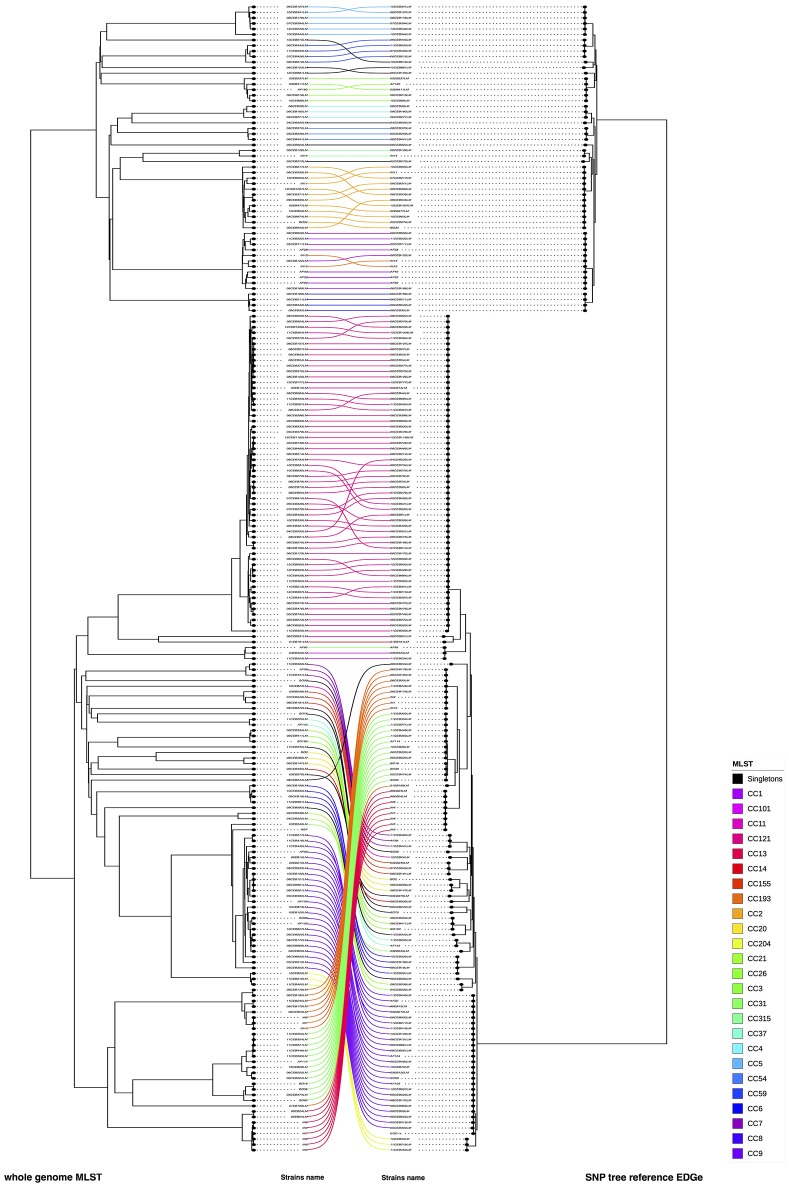
Description:Background/objectives: Whole genome sequencing (WGS) has proven to be a powerful subtyping tool for foodborne pathogenic bacteria like L. monocytogenes. The interests of genome-scale analysis for national surveillance, outbreak detection or source tracking has been largely documented. The genomic data however can be exploited with many different bioinformatics methods like single nucleotide polymorphism (SNP), core-genome multi locus sequence typing (cgMLST), whole-genome multi locus sequence typing (wgMLST) or multi locus predicted protein sequence typing (MLPPST) on either core-genome (cgMLPPST) or pan-genome (wgMLPPST). Currently, there are little comparisons studies of these different analytical approaches. Our objective was to assess and compare different genomic methods that can be implemented in order to cluster isolates of L. monocytogenes. Methods: The clustering methods were evaluated on a collection of 207 L. monocytogenes genomes of food origin representative of the genetic diversity of the Anses collection. The trees were then compared using robust statistical analyses. Results: The backward comparability between conventional typing methods and genomic methods revealed a near-perfect concordance. The importance of selecting a proper reference when calling SNPs was highlighted, although distances between strains remained identical. The analysis also revealed that the topology of the phylogenetic trees between wgMLST and cgMLST were remarkably similar. The comparison between SNP and cgMLST or SNP and wgMLST approaches showed that the topologies of phylogenic trees were statistically similar with an almost equivalent clustering. Conclusion: Our study revealed high concordance between wgMLST, cgMLST, and SNP approaches which are all suitable for typing of L. monocytogenes. The comparable clustering is an important observation considering that the two approaches have been variously implemented among reference laboratories.
-
Subjects:
-
Source:Front Microbiol. 2017; 8.
-
Pubmed ID:29238330
-
Pubmed Central ID:PMC5712588
-
Document Type:
-
Volume:8
-
Collection(s):
-
Main Document Checksum:urn:sha256:0a4451e942b22aa15f6aee3f36e4f821012c7829a034c30320e906c3ad7eafe2
-
Download URL:
-
File Type:
 [PDF
- 4.37 MB
]
[PDF
- 4.37 MB
]
Supporting Files

ON THIS PAGE

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
CDC STACKS serves as an archival repository of CDC-published products including
scientific findings,
journal articles, guidelines, recommendations, or other public health information authored or
co-authored by CDC or funded partners.
As a repository, CDC STACKS retains documents in their original published format to ensure public access to scientific information.
As a repository, CDC STACKS retains documents in their original published format to ensure public access to scientific information.
You May Also Like
COLLECTION
CDC Public Access