Epidemic History and Evolutionary Dynamics of Hepatitis B Virus Infection in Two Remote Communities in Rural Nigeria
Supporting Files
Public Domain

-
Jul 19 2010
-
File Language:
English
Details
-
Alternative Title:PLoS One
-
Personal Author:
-
Description:Background
In Nigeria, hepatitis B virus (HBV) infection has reached hyperendemic levels and its nature and origin have been described as a puzzle. In this study, we investigated the molecular epidemiology and epidemic history of HBV infection in two semi-isolated rural communities in North/Central Nigeria. It was expected that only a few, if any, HBV strains could have been introduced and effectively transmitted among these residents, reflecting limited contacts of these communities with the general population in the country.
Methods and Findings
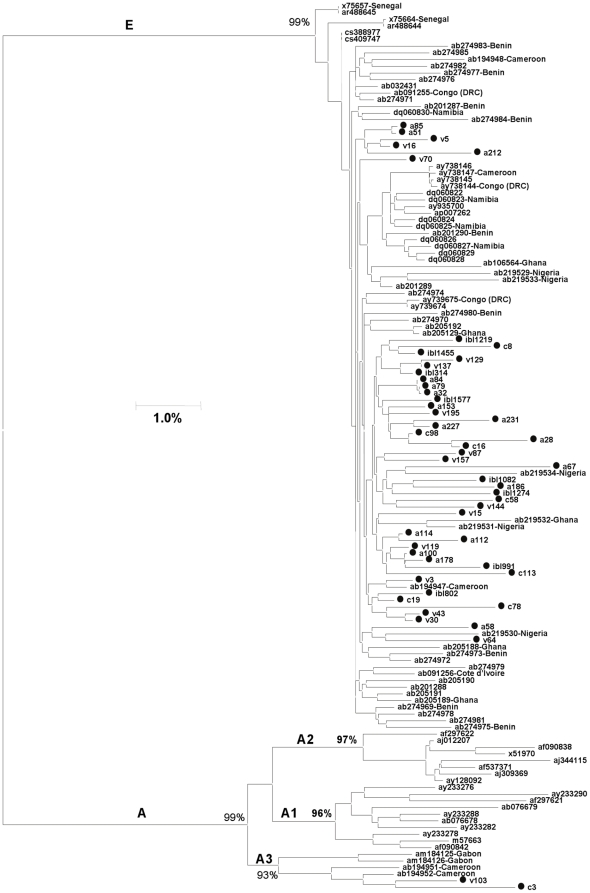
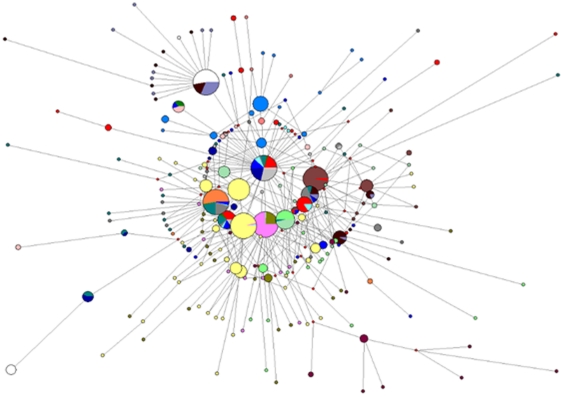
Despite remoteness and isolation, ∼11% of the entire population in these communities was HBV-DNA seropositive. Analyses of the S-gene sequences obtained from 55 HBV-seropositive individuals showed the circulation of 37 distinct HBV variants. These HBV isolates belong predominantly to genotype E (HBV/E) (n = 53, 96.4%), with only 2 classified as sub-genotype A3 (HBV/A3). Phylogenetic analysis showed extensive intermixing between HBV/E variants identified in these communities and different countries in Africa. Quasispecies analysis of 22 HBV/E strains using end-point limiting-dilution real-time PCR, sequencing and median joining networks showed extensive intra-host heterogeneity and inter-host variant sharing. To investigate events that resulted in such remarkable HBV/E diversity, HBV full-size genome sequences were obtained from 47 HBV/E infected persons and P gene was subjected to Bayesian coalescent analysis. The time to the most recent common ancestor (tMRCA) for these HBV/E variants was estimated to be year 1952 (95% highest posterior density (95% HPD): 1927–1970). Using additional HBV/E sequences from other African countries, the tMRCA was estimated to be year 1948 (95% HPD: 1924–1966), indicating that HBV/E in these remote communities has a similar time of origin with multiple HBV/E variants broadly circulating in West/Central Africa. Phylogenetic analysis and statistical neutrality tests suggested rapid HBV/E population expansion. Additionally, skyline plot analysis showed an increase in the size of the HBV/E-infected population over the last ∼30–40 years.
Conclusions
Our data suggest a massive introduction and relatively recent HBV/E expansion in the human population in Africa. Collectively, these data show a significant shift in the HBV/E epidemic dynamics in Africa over the last century.
-
Subjects:
-
Source:PLoS One. 5(7).
-
Document Type:
-
Place as Subject:
-
Volume:5
-
Issue:7
-
Collection(s):
-
Main Document Checksum:urn:sha-512:94f42e3c43f790e01301fea0264ac10b211afa05c26e1d7ac11c3f7225aa486597dd39512598dc42fe3b78862e5c5c355021967d21209948760d09612b8f38da
-
Download URL:
-
File Type:
 [PDF
- 943.86 KB
]
[PDF
- 943.86 KB
]
Supporting Files

File Language:
English
ON THIS PAGE

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
CDC STACKS serves as an archival repository of CDC-published products including
scientific findings,
journal articles, guidelines, recommendations, or other public health information authored or
co-authored by CDC or funded partners.
As a repository, CDC STACKS retains documents in their original published format to ensure public access to scientific information.
As a repository, CDC STACKS retains documents in their original published format to ensure public access to scientific information.
You May Also Like
COLLECTION
CDC Public Access