Genome Structural Diversity among 31 Bordetella pertussis Isolates from Two Recent U.S. Whooping Cough Statewide Epidemics
Supporting Files
Public Domain

-
2016 May-Jun
-
Details
-
Alternative Title:mSphere
-
Personal Author:
-
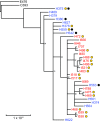
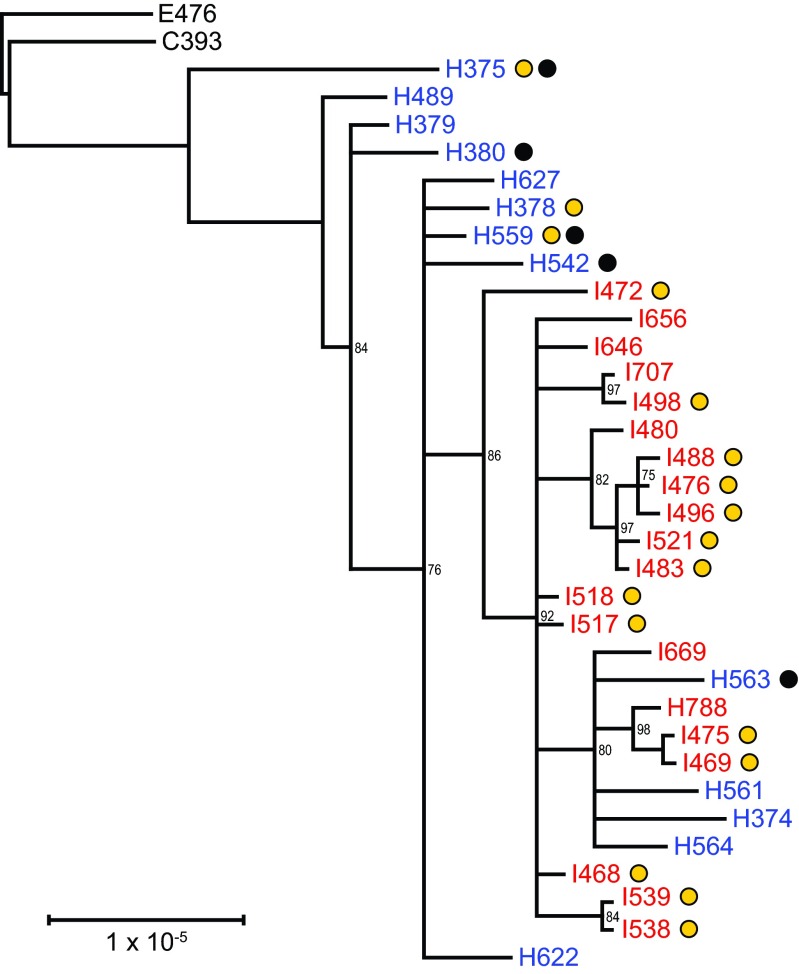
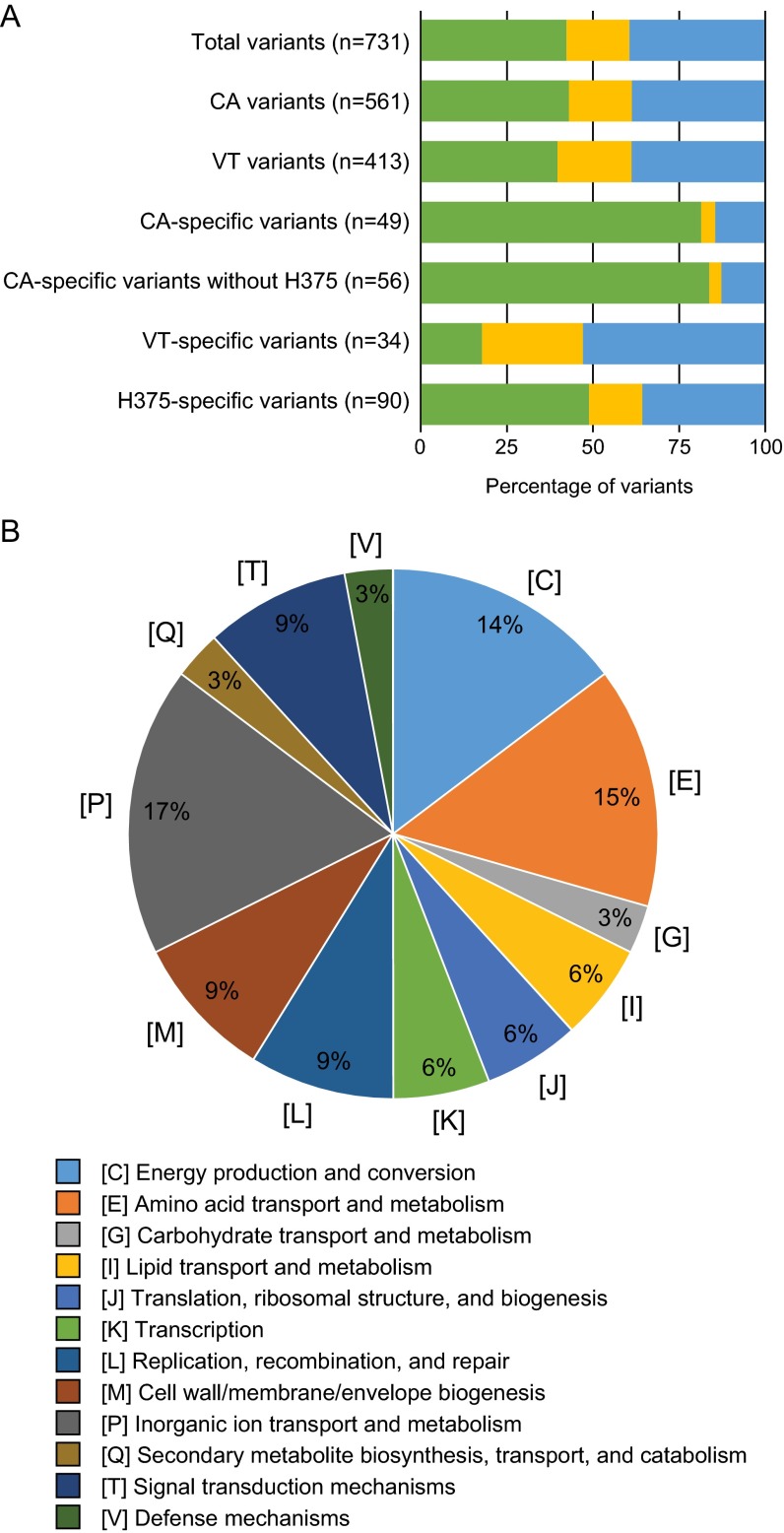
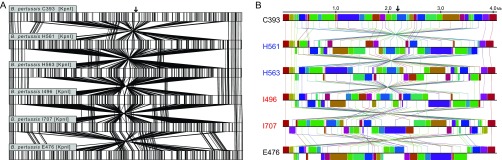
Description:During 2010 and 2012, California and Vermont, respectively, experienced statewide epidemics of pertussis with differences seen in the demographic affected, case clinical presentation, and molecular epidemiology of the circulating strains. To overcome limitations of the current molecular typing methods for pertussis, we utilized whole-genome sequencing to gain a broader understanding of how current circulating strains are causing large epidemics. Through the use of combined next-generation sequencing technologies, this study compared de novo, single-contig genome assemblies from 31 out of 33 Bordetella pertussis isolates collected during two separate pertussis statewide epidemics and 2 resequenced vaccine strains. Final genome architecture assemblies were verified with whole-genome optical mapping. Sixteen distinct genome rearrangement profiles were observed in epidemic isolate genomes, all of which were distinct from the genome structures of the two resequenced vaccine strains. These rearrangements appear to be mediated by repetitive sequence elements, such as high-copy-number mobile genetic elements and rRNA operons. Additionally, novel and previously identified single nucleotide polymorphisms were detected in 10 virulence-related genes in the epidemic isolates. Whole-genome variation analysis identified state-specific variants, and coding regions bearing nonsynonymous mutations were classified into functional annotated orthologous groups. Comprehensive studies on whole genomes are needed to understand the resurgence of pertussis and develop novel tools to better characterize the molecular epidemiology of evolving B. pertussis populations. IMPORTANCE Pertussis, or whooping cough, is the most poorly controlled vaccine-preventable bacterial disease in the United States, which has experienced a resurgence for more than a decade. Once viewed as a monomorphic pathogen, B. pertussis strains circulating during epidemics exhibit diversity visible on a genome structural level, previously undetectable by traditional sequence analysis using short-read technologies. For the first time, we combine short- and long-read sequencing platforms with restriction optical mapping for single-contig, de novo assembly of 31 isolates to investigate two geographically and temporally independent U.S. pertussis epidemics. These complete genomes reshape our understanding of B. pertussis evolution and strengthen molecular epidemiology toward one day understanding the resurgence of pertussis.
-
Subjects:
-
Source:mSphere. 2016; 1(3).
-
Pubmed ID:27303739
-
Pubmed Central ID:PMC4888882
-
Document Type:
-
Volume:1
-
Issue:3
-
Collection(s):
-
Main Document Checksum:urn:sha256:fc1a6d75f3d788480dc5bdc2e93dc6c234be5926e64a75ad9ca3ae9ef7075e3e
-
Download URL:
-
File Type:
 [PDF
- 717.42 KB
]
[PDF
- 717.42 KB
]
Supporting Files

ON THIS PAGE

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
CDC STACKS serves as an archival repository of CDC-published products including
scientific findings,
journal articles, guidelines, recommendations, or other public health information authored or
co-authored by CDC or funded partners.
As a repository, CDC STACKS retains documents in their original published format to ensure public access to scientific information.
As a repository, CDC STACKS retains documents in their original published format to ensure public access to scientific information.
You May Also Like
COLLECTION
CDC Public Access