Canonical Single Nucleotide Polymorphisms (SNPs) for High-Resolution Subtyping of Shiga-Toxin Producing Escherichia coli (STEC) O157:H7
Supporting Files
Public Domain

-
Jul 01 2015
-
Details
-
Alternative Title:PLoS One
-
Personal Author:
-
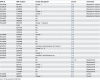
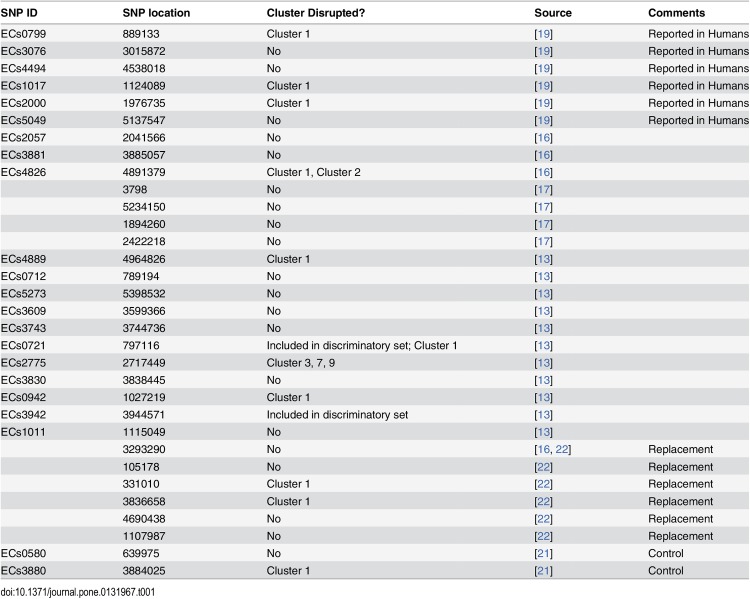
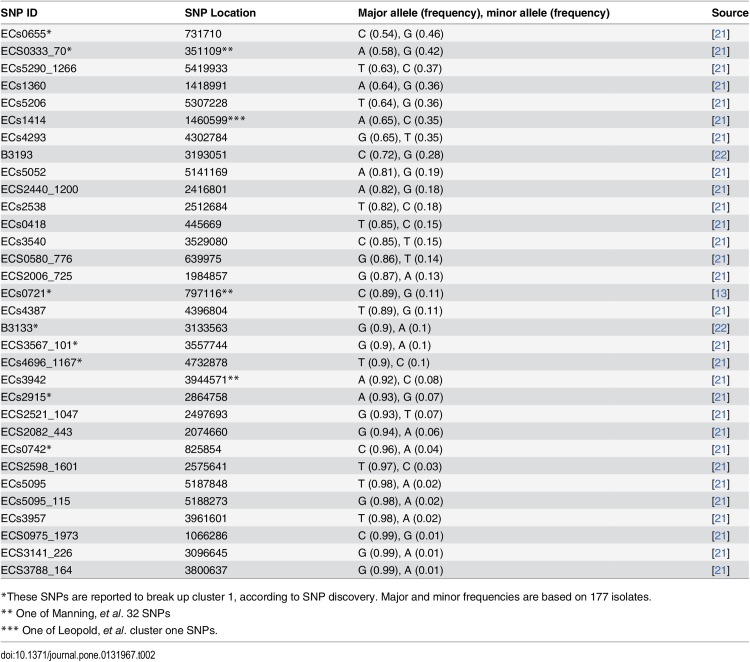
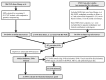
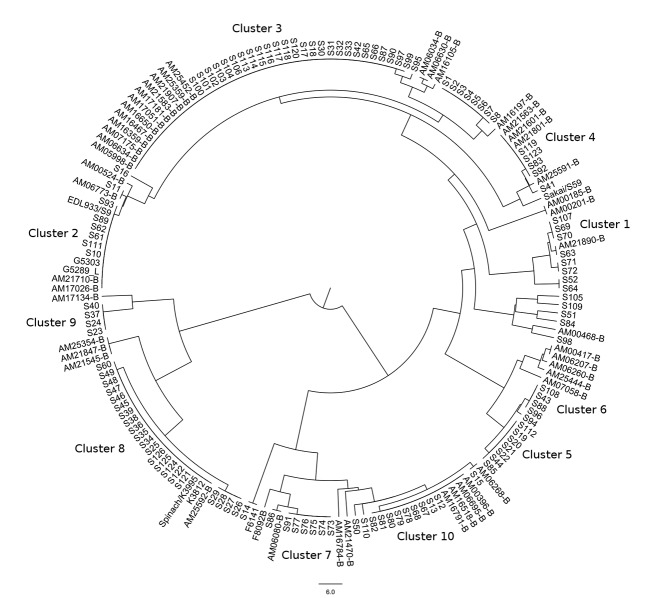
Description:The objective of this study was to develop a canonical, parsimoniously-informative SNP panel for subtyping Shiga-toxin producing Escherichia coli (STEC) O157:H7 that would be consistent with epidemiological, PFGE, and MLVA clustering of human specimens. Our group had previously identified 906 putative discriminatory SNPs, which were pared down to 391 SNPs based on their prevalence in a test set. The 391 SNPs were screened using a high-throughput form of TaqMan PCR against a set of clinical isolates that represent the most diverse collection of O157:H7 isolates from outbreaks and sporadic cases examined to date. Another 30 SNPs identified by others were also screened using the same method. Two additional targets were tested using standard TaqMan PCR endpoint analysis. These 423 SNPs were reduced to a 32 SNP panel with the almost the same discriminatory value. While the panel partitioned our diverse set of isolates in a manner that was consistent with epidemiological data and PFGE and MLVA phylogenies, it resulted in fewer subtypes than either existing method and insufficient epidemiological resolution in 10 of 47 clusters. Therefore, another round of SNP discovery was undertaken using comparative genomic resequencing of pooled DNA from the 10 clusters with insufficient resolution. This process identified 4,040 potential SNPs and suggested one of the ten clusters was incorrectly grouped. After its removal, there were 2,878 SNPs, of which only 63 were previously identified and 438 occurred across multiple clusters. Among highly clonal bacteria like STEC O157:H7, linkage disequilibrium greatly limits the number of parsimoniously informative SNPs. Therefore, it is perhaps unsurprising that our panel accounted for the potential discriminatory value of numerous other SNPs reported in the literature. We concluded published O157:H7 SNPs are insufficient for effective epidemiological subtyping. However, the 438 multi-cluster SNPs we identified may provide the additional information required.
-
Subjects:
-
Source:PLoS One. 10(7).
-
Pubmed ID:26132731
-
Pubmed Central ID:PMC4488506
-
Document Type:
-
Volume:10
-
Issue:7
-
Collection(s):
-
Main Document Checksum:urn:sha256:b18da9bae319898cb230b4d5314b4358d4a66118725a95b43df31043c813222a
-
Download URL:
-
File Type:
 [PDF
- 582.11 KB
]
[PDF
- 582.11 KB
]
Supporting Files

ON THIS PAGE

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
CDC STACKS serves as an archival repository of CDC-published products including
scientific findings,
journal articles, guidelines, recommendations, or other public health information authored or
co-authored by CDC or funded partners.
As a repository, CDC STACKS retains documents in their original published format to ensure public access to scientific information.
As a repository, CDC STACKS retains documents in their original published format to ensure public access to scientific information.
You May Also Like
COLLECTION
CDC Public Access