The mathematical limits of genetic prediction for complex chronic disease
Supporting Files

-
Feb 03 2015
-
Details
-
Alternative Title:J Epidemiol Community Health
-
Personal Author:
-
Description:Background
Attempts at predicting individual risk of disease based on common germline genetic variation have largely been disappointing. The present paper formalises why genetic prediction at the individual level is and will continue to have limited utility given the aetiological architecture of most common complex diseases.
Methods
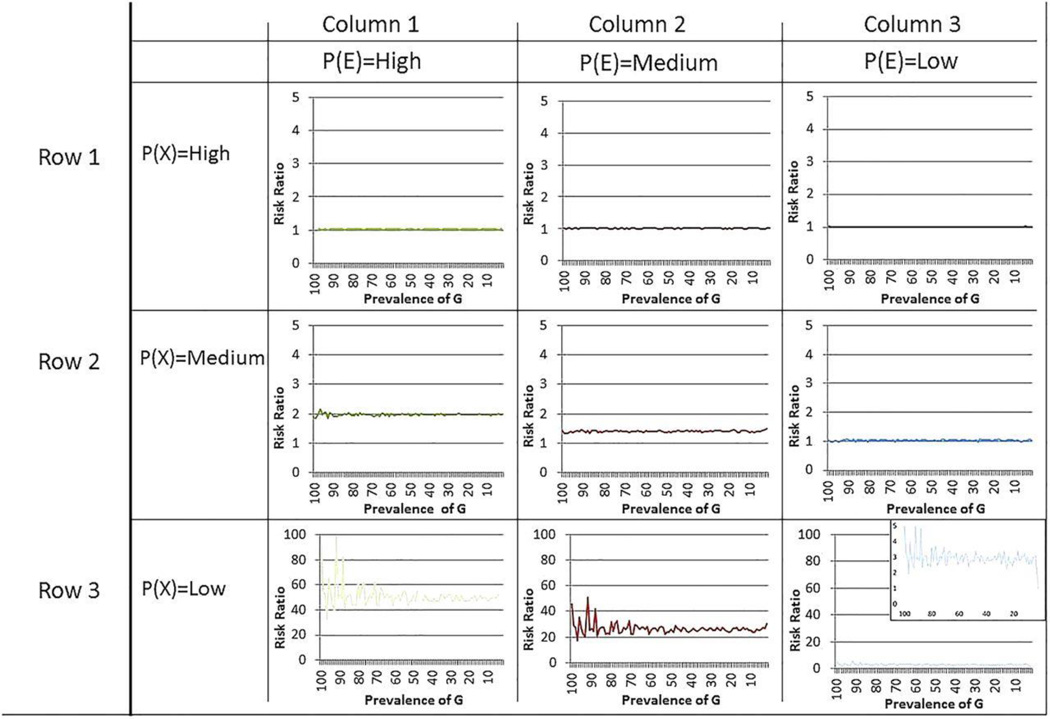
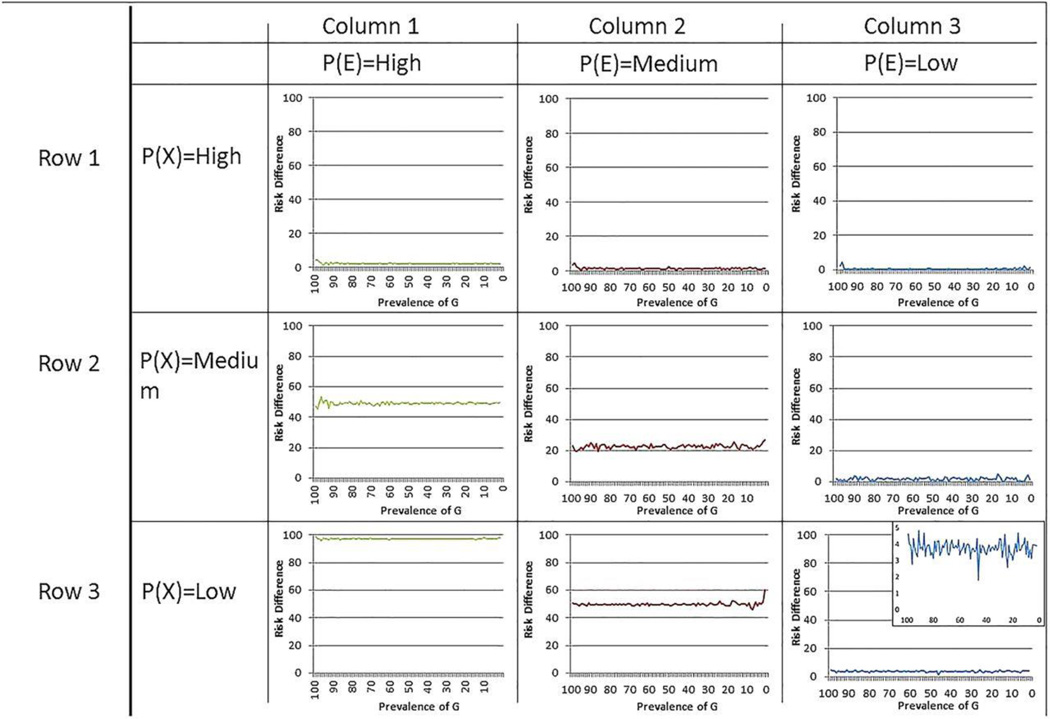
Data were simulated on one million populations with 10 000 individuals in each populations with varying prevalences of a genetic risk factor, an interacting environmental factor and the background rate of disease. The determinant risk ratio and risk difference magnitude for the association between a gene variant and disease is a function of the prevalence of the interacting factors that activate the gene, and the background rate of disease.
Results
The risk ratio and total excess cases due to the genetic factor increase as the prevalence of interacting factors increase, and decrease as the background rate of disease increases. Germline genetic variations have high predictive capacity for individual disease only under conditions of high heritability of particular genetic sequences, plausible only under rare variant hypotheses.
Conclusions
Under a model of common germline genetic variants that interact with other genes and/or environmental factors in order to cause disease, the predictive capacity of common genetic variants is determined by the prevalence of the factors that interact with the variant and the background rate. A focus on estimating genetic associations for the purpose of prediction without explicitly grounding such work in an understanding of modifiable (including environmentally influenced) factors will be limited in its ability to yield important insights about the risk of disease.
-
Subjects:
-
Source:J Epidemiol Community Health. 2015; 69(6):574-579.
-
Pubmed ID:25648993
-
Pubmed Central ID:PMC4430395
-
Document Type:
-
Funding:DA034244/DA/NIDA NIH HHS/United States ; K01 AA021511/AA/NIAAA NIH HHS/United States ; K01-AA021511/AA/NIAAA NIH HHS/United States ; MH 082598/MH/NIMH NIH HHS/United States ; MH 082729/MH/NIMH NIH HHS/United States ; MH 095718/MH/NIMH NIH HHS/United States ; P51RR000165/RR/NCRR NIH HHS/United States ; R01 MH082729/MH/NIMH NIH HHS/United States ; R01 MH093612/MH/NIMH NIH HHS/United States ; R01MH093612/MH/NIMH NIH HHS/United States ; RC4MH092707/MH/NIMH NIH HHS/United States ; U01OH010407/OH/NIOSH CDC HHS/United States ; U01OH010416/OH/NIOSH CDC HHS/United States ; Medical Research Council/United Kingdom
-
Volume:69
-
Issue:6
-
Collection(s):
-
Main Document Checksum:urn:sha256:d4a02d2ad7f3c92dc24285194e99cf516da6a0604e8bb0df30be0f4d08383e3e
-
Download URL:
-
File Type:
 [PDF
- 730.74 KB
]
[PDF
- 730.74 KB
]
Supporting Files

ON THIS PAGE

{kind=link}
{kind=link}
{kind=link}
{kind=link}
CDC STACKS serves as an archival repository of CDC-published products including
scientific findings,
journal articles, guidelines, recommendations, or other public health information authored or
co-authored by CDC or funded partners.
As a repository, CDC STACKS retains documents in their original published format to ensure public access to scientific information.
As a repository, CDC STACKS retains documents in their original published format to ensure public access to scientific information.
You May Also Like
COLLECTION
CDC Public Access