Distinct pathological phenotypes of Creutzfeldt-Jakob disease in recipients of prion-contaminated growth hormone
Supporting Files
Public Domain

-
Jun 25 2015
-
Details
-
Alternative Title:Acta Neuropathol Commun
-
Personal Author:
-
Description:Introduction
The present study compares the clinical, pathological and molecular features of a United States (US) case of growth hormone (GH)-associated Creutzfeldt-Jakob disease (GH-CJD) (index case) to those of two earlier referred US cases of GH-CJD and one case of dura mater (d)-associated CJD (dCJD). All iatrogenic CJD (iCJD) subjects were methionine (M) homozygous at codon 129 (129MM) of the prion protein (PrP) gene and had scrapie prion protein (PrPSc) type 1 (iCJDMM1).
Results
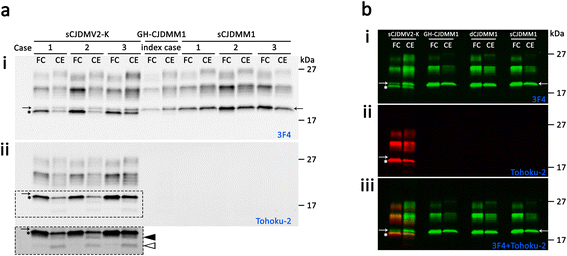
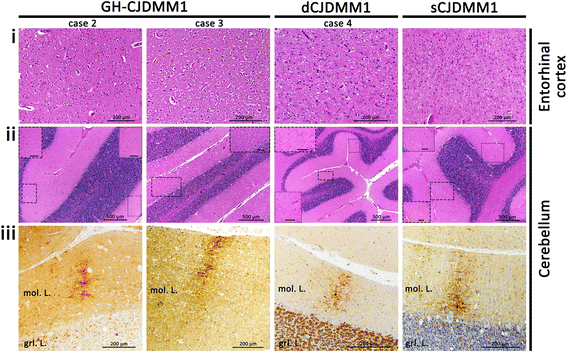
The index subject presented with ataxia, weight loss and changes in the sleep pattern about 38 years after the midpoint of GH treatment. Autopsy examination revealed a neuropathological phenotype reminiscent of both sCJDMV2-K (a sporadic CJD subtype in subjects methionine/valine heterozygous at codon 129 with PrPSc type 2 and the presence of kuru plaques) and variant CJD (vCJD). The two earlier cases of GH-CJDMM1 and the one of dCJDMM1 were associated with neuropathological phenotypes that differed from that of the index case mainly because they lacked PrP plaques. The phenotype of the earlier GH-CJDMM1 cases shared several, but not all, characteristics with sCJDMM1, whereas dCJDMM1 was phenotypically indistinguishable from sCJDMM1. Two distinct groups of dCJDMM1 have also been described in Japan based on clinical features, the presence or absence of PrP plaques and distinct PK-resistant PrPSc (resPrPSc) electrophoretic mobilities. The resPrPSc electrophoretic mobility was, however, identical in our GH-CJDMM1 and dCJDMM1 cases, and matched that of sCJDMM1.
Conclusions
Our study shows that receipt of prion-contaminated GH can lead to a prion disease with molecular features (129MM and PrPSc type 2) and phenotypic characteristics that differ from those of sporadic prion disease (sCJDMM1), a difference that may reflect adaptation of “heterologous” prion strains to the 129MM background.
-
Subjects:
-
Source:Acta Neuropathol Commun. 3.
-
Pubmed ID:26108478
-
Pubmed Central ID:PMC4479081
-
Document Type:
-
Volume:3
-
Collection(s):
-
Main Document Checksum:urn:sha256:a85a576f8dd6f102ef6baad95d1afd40b87a92c461b016d176103bbea0e424d9
-
Download URL:
-
File Type:
 [PDF
- 1.62 MB
]
[PDF
- 1.62 MB
]
Supporting Files

ON THIS PAGE

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
CDC STACKS serves as an archival repository of CDC-published products including
scientific findings,
journal articles, guidelines, recommendations, or other public health information authored or
co-authored by CDC or funded partners.
As a repository, CDC STACKS retains documents in their original published format to ensure public access to scientific information.
As a repository, CDC STACKS retains documents in their original published format to ensure public access to scientific information.
You May Also Like
COLLECTION
CDC Public Access