Sodium Current Reduction Unmasks a Structure-Dependent Substrate for Arrhythmogenesis in the Normal Ventricles
Supporting Files

-
Jan 28 2014
-
File Language:
English
Details
-
Alternative Title:PLoS One
-
Personal Author:
-
Description:Background
Organ-scale arrhythmogenic consequences of source-sink mismatch caused by impaired excitability remain unknown, hindering the understanding of pathophysiology in disease states like Brugada syndrome and ischemia.
Objective
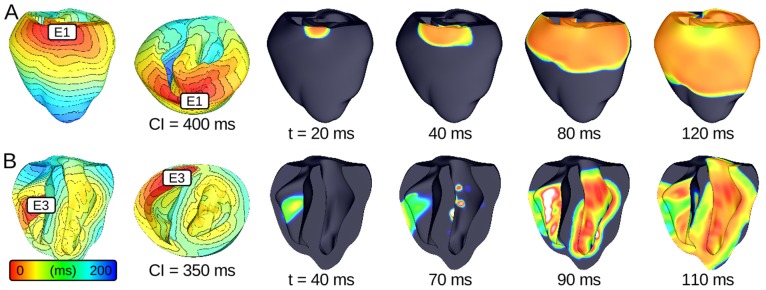
We sought to determine whether sodium current (INa) reduction in the structurally normal heart unmasks a regionally heterogeneous substrate for the induction of sustained arrhythmia by premature ventricular contractions (PVCs).
Methods
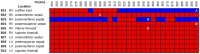
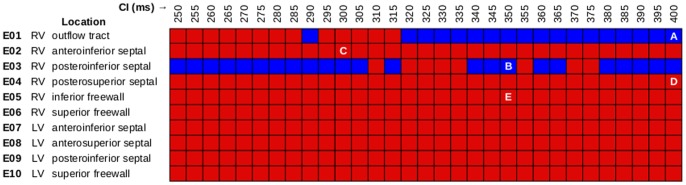
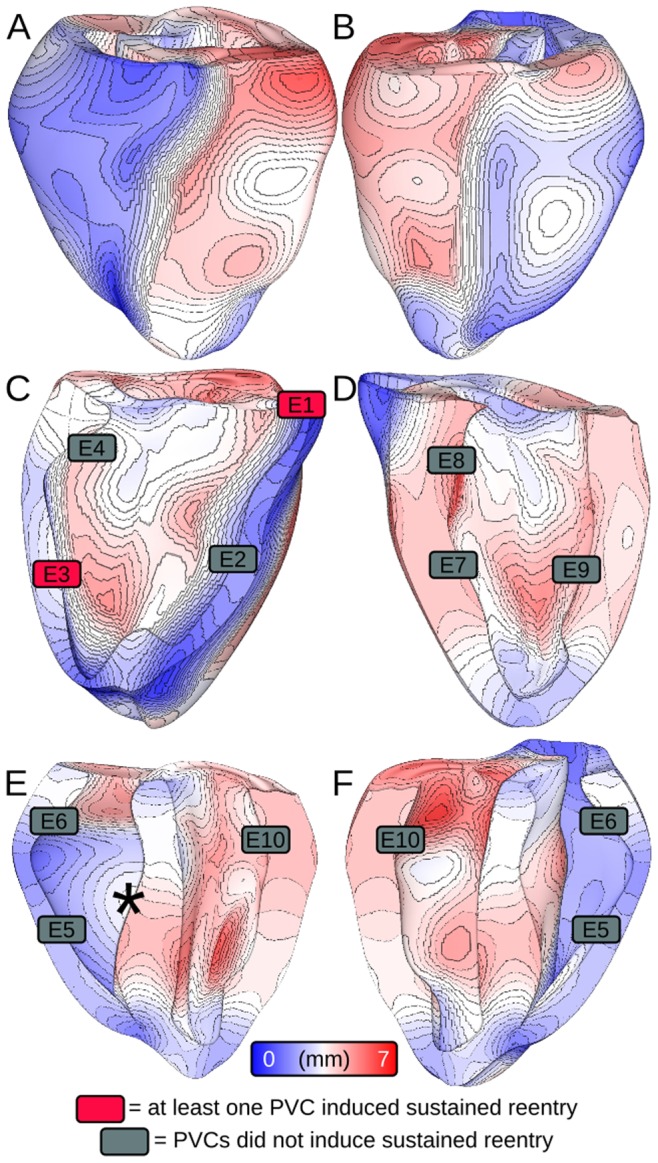
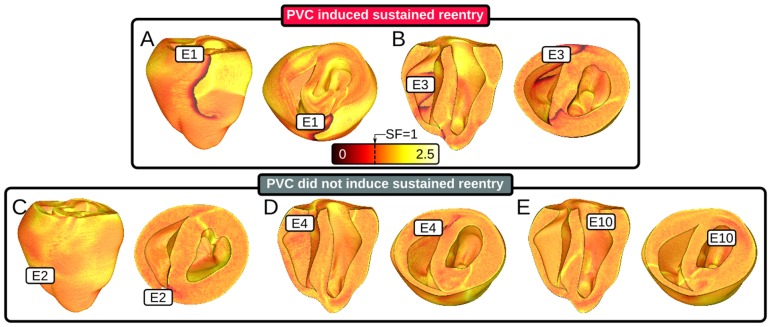
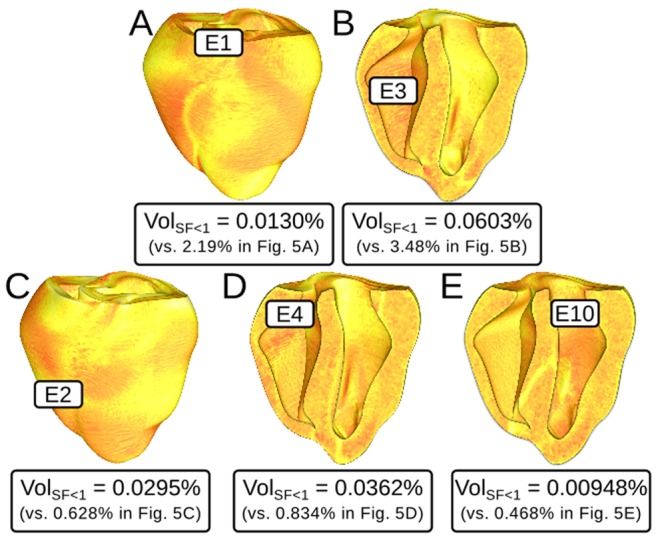
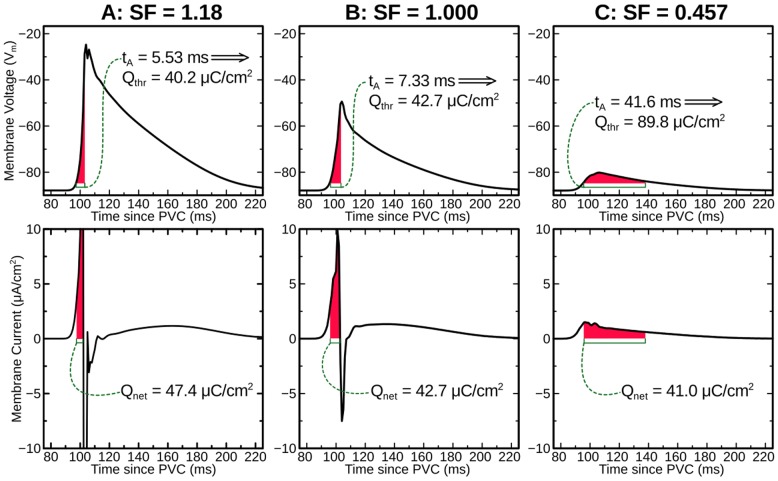
We conducted simulations in rabbit ventricular computer models with 930 unique combinations of PVC location (10 sites) and coupling interval (250–400 ms), INa reduction (30 or 40% of normal levels), and post-PVC sinus rhythm (arrested or persistent). Geometric characteristics and source-sink mismatch were quantitatively analyzed by calculating ventricular wall thickness and a newly formulated 3D safety factor (SF), respectively.
Results
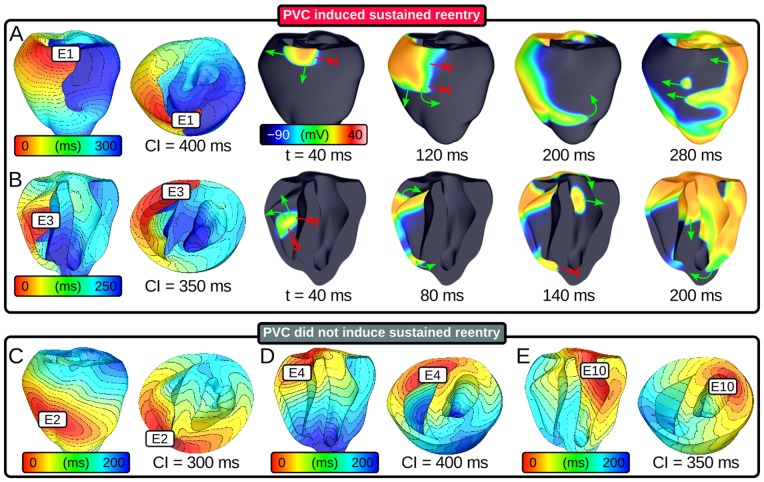
Reducing INa to 30% of its normal level created a substrate for sustained arrhythmia induction by establishing large regions of critical source-sink mismatch (SF<1) for ectopic wavefronts propagating from thin to thick tissue. In the same simulations but with 40% of normal INa, PVCs did not induce reentry because the volume of tissue with SF<1 was >95% smaller. Likewise, when post-PVC sinus activations were persistent instead of arrested, no ectopic excitations initiated sustained reentry because sinus activation breakthroughs engulfed the excitable gap.
Conclusion
Our new SF formulation can quantify ectopic wavefront propagation robustness in geometrically complex 3D tissue with impaired excitability. This novel methodology was applied to show that INa reduction precipitates source-sink mismatch, creating a potent substrate for sustained arrhythmia induction by PVCs originating near regions of ventricular wall expansion, such as the RV outflow tract.
-
Subjects:
-
Source:PLoS One. 2014; 9(1).
-
Pubmed ID:24489810
-
Pubmed Central ID:PMC3904970
-
Document Type:
-
Funding:
-
Volume:9
-
Issue:1
-
Collection(s):
-
Main Document Checksum:urn:sha256:bd3fb6a6a8a0e039fecfb3f19fd66988aced5fb1f155fb478e0afa17c5d64313
-
Download URL:
-
File Type:
 [PDF
- 1.31 MB
]
[PDF
- 1.31 MB
]
Supporting Files

File Language:
English
ON THIS PAGE

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
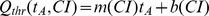{kind=link}
{kind=link}
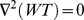{kind=link}
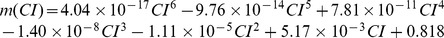{kind=link}
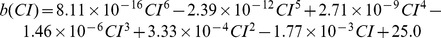{kind=link}
{kind=link}
{kind=link}
CDC STACKS serves as an archival repository of CDC-published products including
scientific findings,
journal articles, guidelines, recommendations, or other public health information authored or
co-authored by CDC or funded partners.
As a repository, CDC STACKS retains documents in their original published format to ensure public access to scientific information.
As a repository, CDC STACKS retains documents in their original published format to ensure public access to scientific information.
You May Also Like
COLLECTION
CDC Public Access