Deep-Sequencing of the Peach Latent Mosaic Viroid Reveals New Aspects of Population Heterogeneity
Supporting Files
Public Domain

-
Jan 30 2014
-
File Language:
English
Details
-
Alternative Title:PLoS One
-
Personal Author:
-
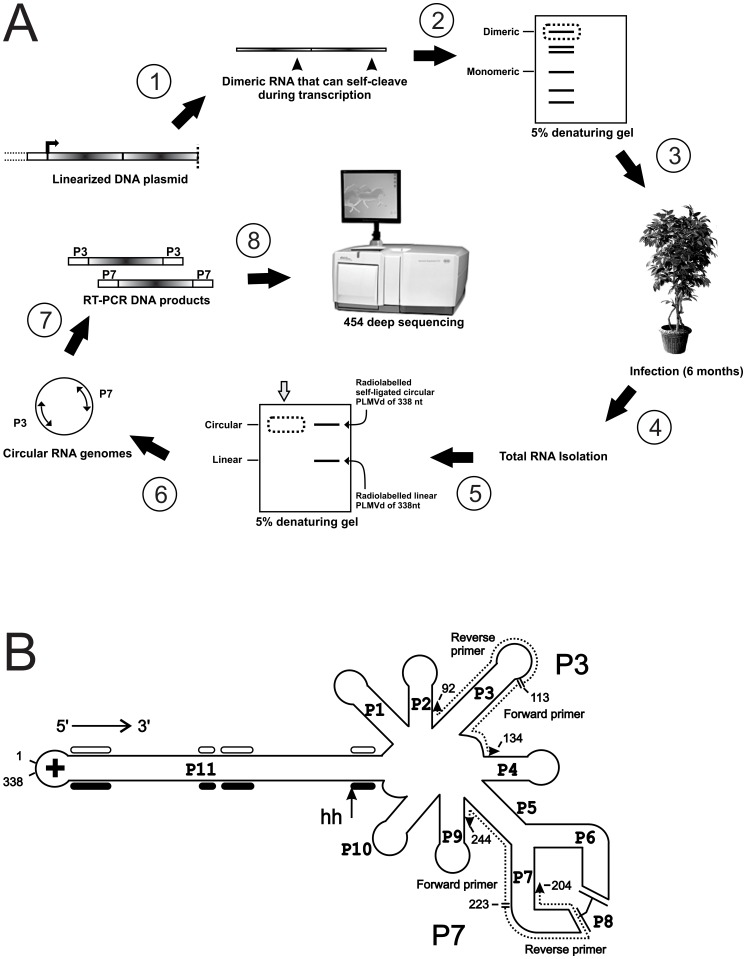
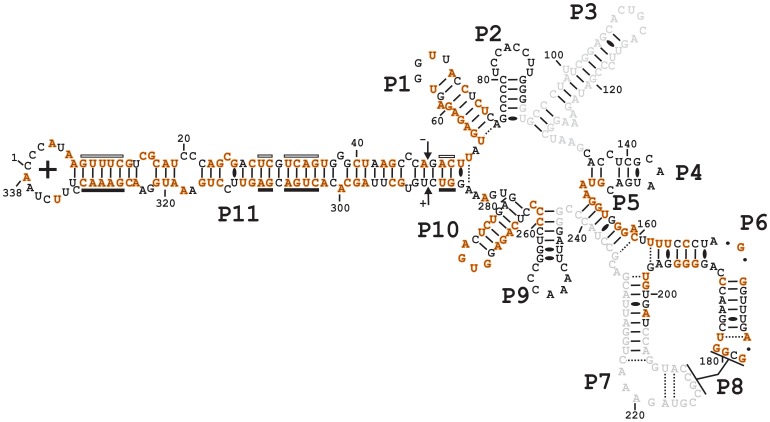
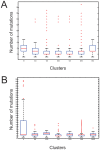
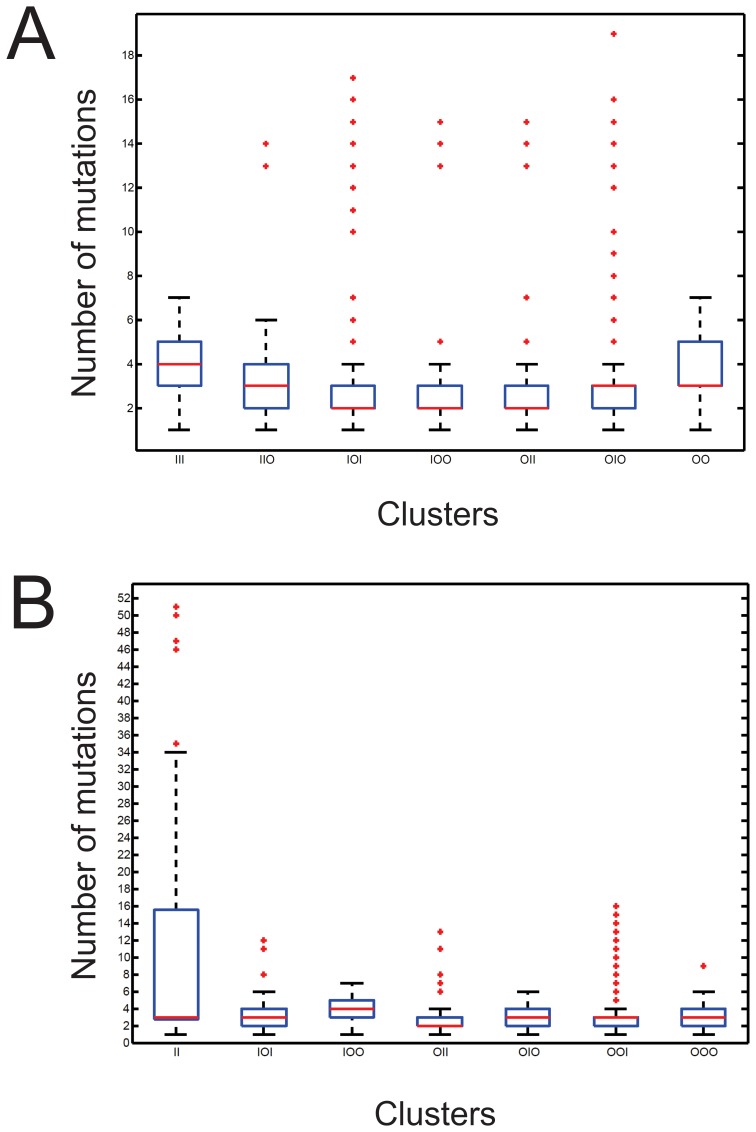
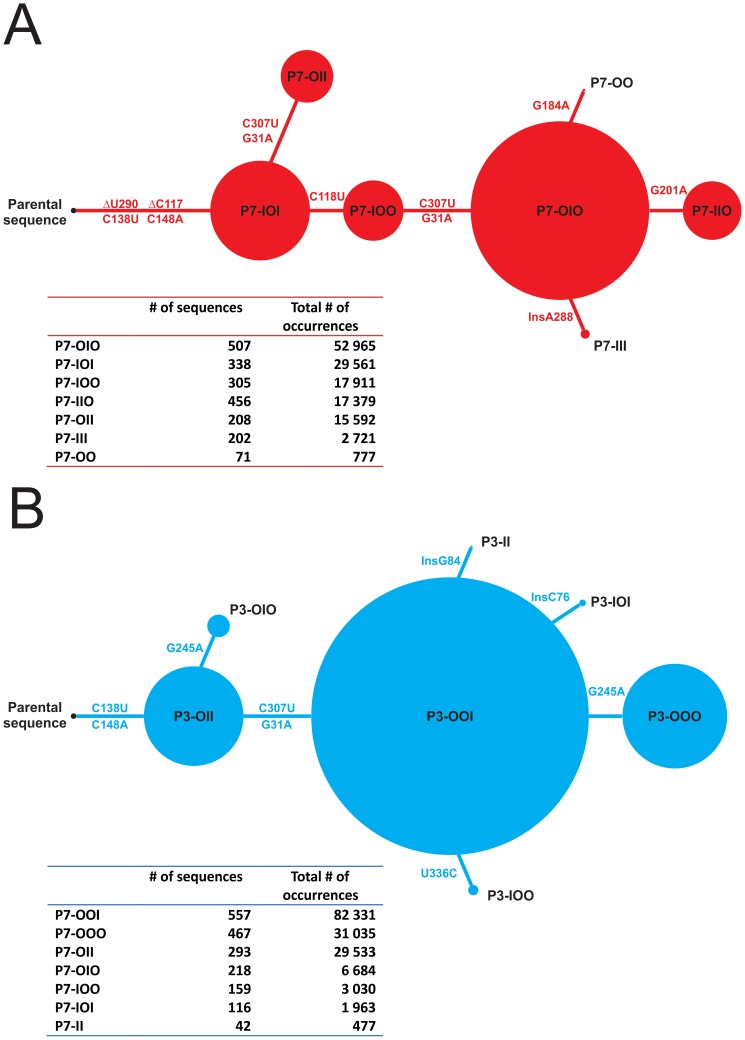
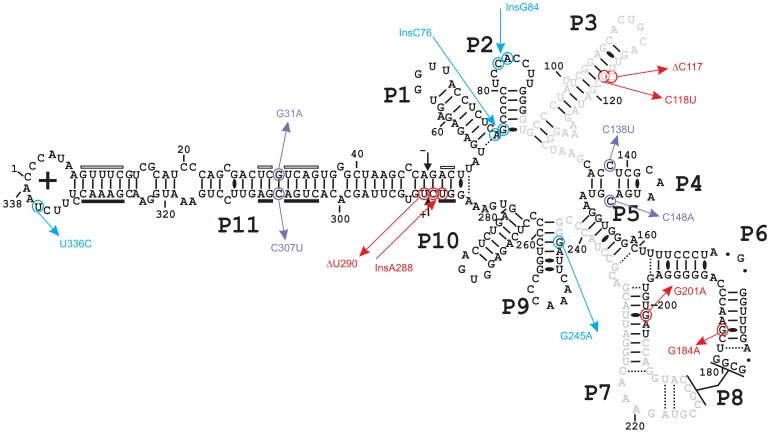
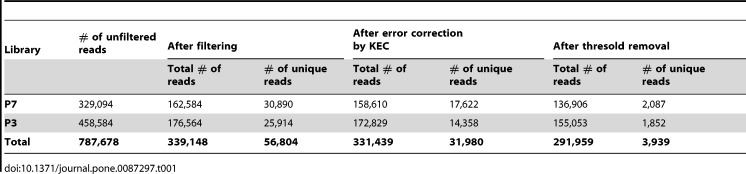
Description:Viroids are small circular single-stranded infectious RNAs characterized by a relatively high mutation level. Knowledge of their sequence heterogeneity remains largely elusive and previous studies, using Sanger sequencing, were based on a limited number of sequences. In an attempt to address sequence heterogeneity from a population dynamics perspective, a GF305-indicator peach tree was infected with a single variant of the Avsunviroidae family member Peach latent mosaic viroid (PLMVd). Six months post-inoculation, full-length circular conformers of PLMVd were isolated and deep-sequenced. We devised an original approach to the bioinformatics refinement of our sequence libraries involving important phenotypic data, based on the systematic analysis of hammerhead self-cleavage activity. Two distinct libraries yielded a total of 3,939 different PLMVd variants. Sequence variants exhibiting up to ∼17% of mutations relative to the inoculated viroid were retrieved, clearly illustrating the high level of divergence dynamics within a unique population. While we initially assumed that most positions of the viroid sequence would mutate, we were surprised to discover that ∼50% of positions remained perfectly conserved, including several small stretches as well as a small motif reminiscent of a GNRA tetraloop which are the result of various selective pressures. Using a hierarchical clustering algorithm, the different variants harvested were subdivided into 7 clusters. We found that most sequences contained an average of 4.6 to 6.4 mutations compared to the variant used to initially inoculate the plant. Interestingly, it was possible to reconstitute and compare the sequence evolution of each of these clusters. In doing so, we identified several key mutations. This study provides a reliable pipeline for the treatment of viroid deep-sequencing. It also sheds new light on the extent of sequence variation that a viroid population can sustain, and which may give rise to a quasispecies.
-
Subjects:
-
Source:PLoS One. 2014; 9(1).
-
Document Type:
-
Volume:9
-
Issue:1
-
Collection(s):
-
Main Document Checksum:urn:sha256:6c30da314f6b341b1784cc44a94079a092c84426f3ca690e25d83cd853cd213a
-
Download URL:
-
File Type:
 [PDF
- 2.23 MB
]
[PDF
- 2.23 MB
]
Supporting Files

File Language:
English
ON THIS PAGE

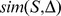{kind=link}
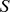{kind=link}
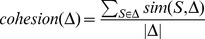{kind=link}
{kind=link}
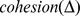{kind=link}
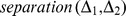{kind=link}
{kind=link}
{kind=link}
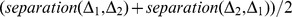{kind=link}
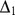{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
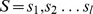{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
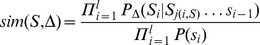{kind=link}
{kind=link}
{kind=link}
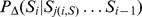{kind=link}
{kind=link}
{kind=link}
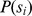{kind=link}
{kind=link}
CDC STACKS serves as an archival repository of CDC-published products including
scientific findings,
journal articles, guidelines, recommendations, or other public health information authored or
co-authored by CDC or funded partners.
As a repository, CDC STACKS retains documents in their original published format to ensure public access to scientific information.
As a repository, CDC STACKS retains documents in their original published format to ensure public access to scientific information.
You May Also Like
COLLECTION
CDC Public Access