Single-cell RNA-seq reveals cell type–specific molecular and genetic associations to lupus
Supporting Files

-
4 08 2022
-
File Language:
English
Details
-
Alternative Title:Science
-
Personal Author:Perez, Richard K. ; Gordon, M. Grace ; Subramaniam, Meena ; Kim, Min Cheol ; Hartoularos, George C. ; Targ, Sasha ; Sun, Yang ; Ogorodnikov, Anton ; Bueno, Raymund ; Lu, Andrew ; Thompson, Mike ; Rappoport, Nadav ; Dahl, Andrew ; Lanata, Cristina M. ; Matloubian, Mehrdad ; Maliskova, Lenka ; Kwek, Serena S. ; Li, Tony ; Slyper, Michal ; Waldman, Julia ; Dionne, Danielle ; Rozenblatt-Rosen, Orit ; Fong, Lawrence ; Dall’Era, Maria ; Balliu, Brunilda ; Regev, Aviv ; Yazdany, Jinoos ; Criswell, Lindsey A. ; Zaitlen, Noah ; Ye, Chun Jimmie
-
Description:INTRODUCTION:
Systemic lupus erythematosus (SLE) is a heterogeneous autoimmune disease with elevated prevalence in women and individuals of Asian, African, and Hispanic ancestry. Bulk transcriptomic profiling has implicated increased type 1 interferon signaling, dysregulated lymphocyte activation, and failure of apoptotic clearance as hallmarks of disease. Many genes participating in these processes are proximal to the ~100 loci associated with SLE. Despite this progress, a comprehensive census of circulating immune cells in SLE remains incomplete, and annotating the cell types and contexts that mediate genetic associations remains challenging.
RATIONALE:
Historically, flow cytometry and bulk transcriptomic analyses were used to profile the composition and gene expression of circulating immune cells in SLE. However, flow cytometry is biased by its use of a limited set of known markers, whereas bulk transcriptomic profiling does not have sufficient power to detect cell type–specific expression differences. Single-cell RNA sequencing (scRNA-seq) of peripheral blood mononuclear cells (PBMCs) holds potential as a comprehensive and unbiased approach to simultaneously profile the composition and transcriptional states of circulating immune cells. However, application of scRNA-seq to population cohorts has been limited by sample throughput, cost, and susceptibility to technical variability. To overcome these limitations, we previously developed multiplexed scRNA-seq (mux-seq) to enable systematic and cost-effective scRNA-seq of population cohorts.
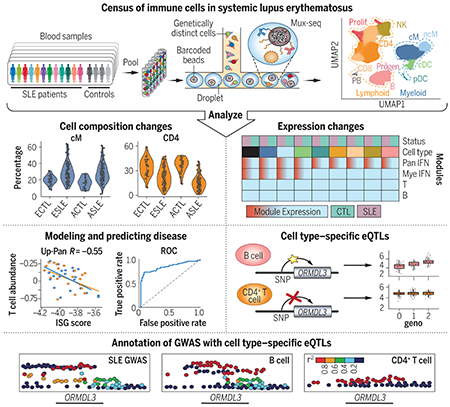
RESULTS:
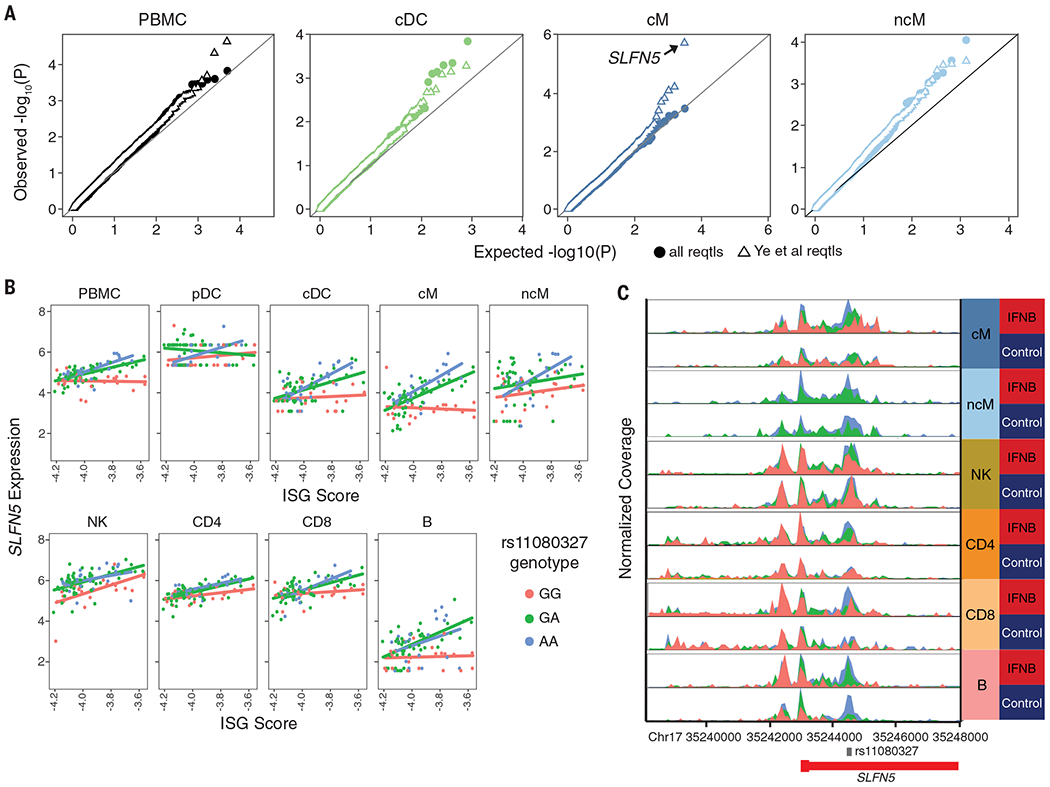
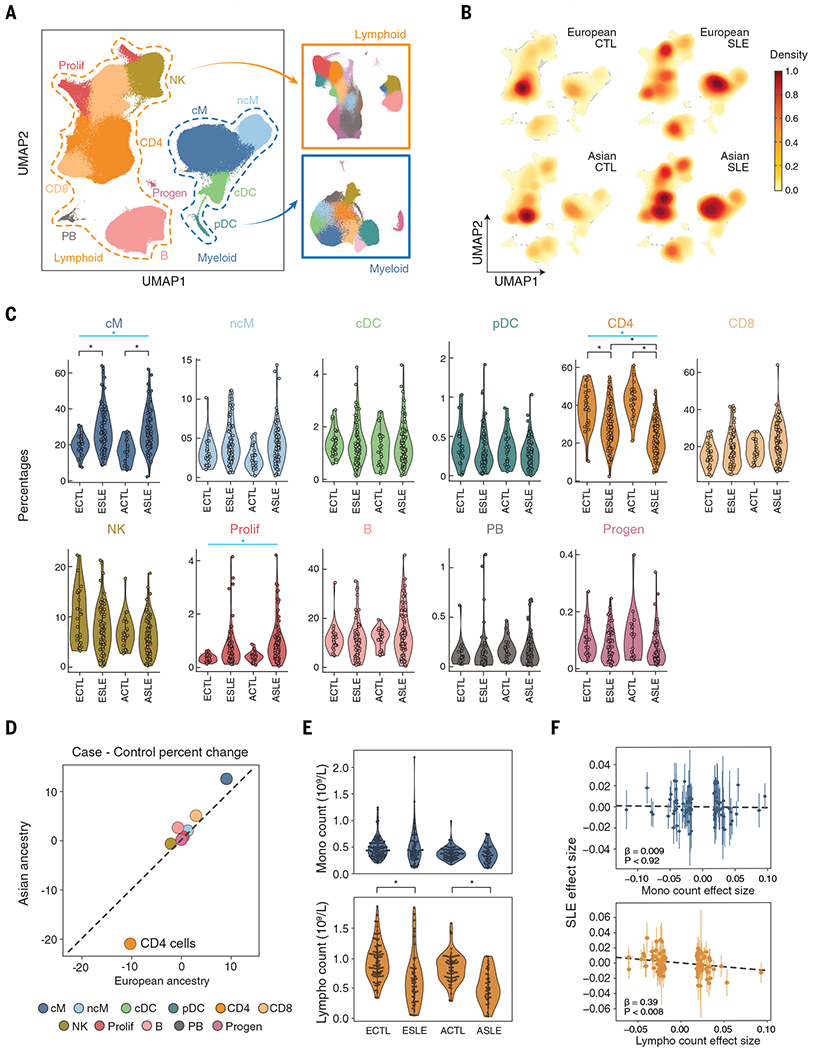
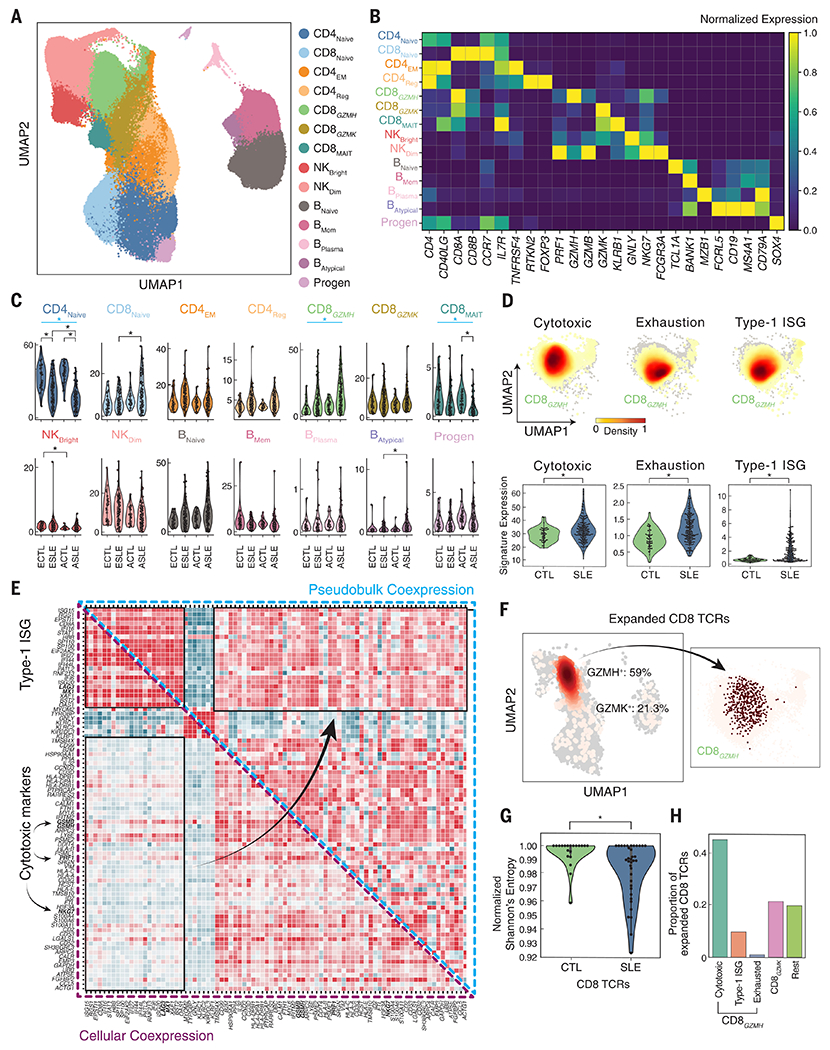
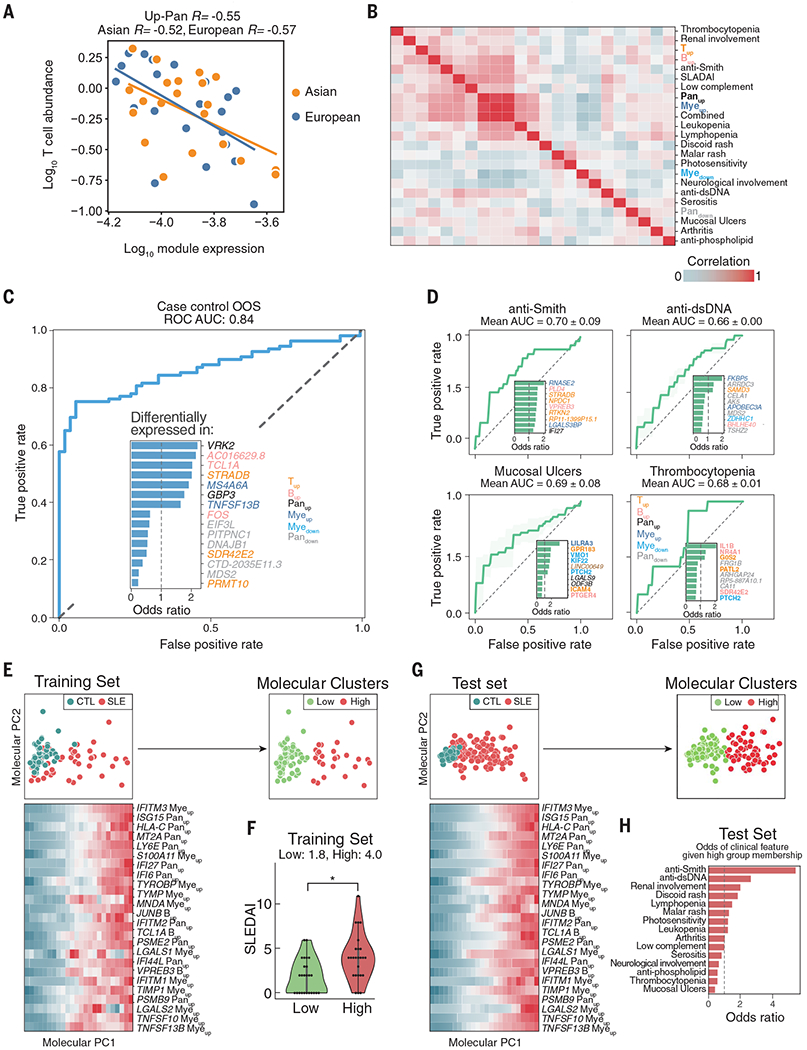
We used mux-seq to profile more than 1.2 million PBMCs from 162 SLE cases and 99 healthy controls of either Asian or European ancestry. SLE cases exhibited differences in both the composition and state of PBMCs. Analysis of lymphocyte composition revealed a reduction in naïve CD4+ T cells and an increase in repertoire-restricted GZMH+ CD8+ T cells. Analysis of transcriptomic profiles across eight cell types revealed that classical monocytes expressed the highest levels of both pan–cell type and myeloid-specific type 1 interferon-stimulated genes (ISGs). The expression of ISGs in monocytes was inversely correlated with naïve CD4+ T cell abundance. Cell type–specific expression features accurately predicted case-control status and stratified patients into molecular subtypes. By integrating genotyping data and using a novel matrix decomposition method, we mapped shared and cell type–specific cis–expression quantitative trait loci (cis-eQTLs) across eight cell types. Cell type–specific cis-eQTLs were enriched for regions of open chromatin specific to the same or related cell types. Joint analysis of cis-eQTLs and genome-wide association study results enabled identification of cell types relevant to immune-mediated diseases, fine-mapping of disease-associated loci, and discovery of novel SLE associations. Interaction analysis identified variants whose effects on gene expression are further modified by interferon activation across patients.
CONCLUSION:
SLE remains challenging to diagnose and treat. The heterogeneity of disease manifestations and treatment response highlight the need for improved molecular characterization. In a large multiethnic cohort, we demonstrate mux-seq as a systematic approach to characterize cellular composition, identify cell type–specific transcriptomic signatures, and annotate genetic variants associated with SLE.
-
Subjects:
-
Source:Science. 376(6589):eabf1970
-
Pubmed ID:35389781
-
Pubmed Central ID:PMC9297655
-
Document Type:
-
Funding:K24 AR074534/AR/NIAMS NIH HHSUnited States/ ; R01 CA227237/CA/NCI NIH HHSUnited States/ ; U01 HG012079/HG/NHGRI NIH HHSUnited States/ ; R03 DE025665/DE/NIDCR NIH HHSUnited States/ ; R01 GM142112/GM/NIGMS NIH HHSUnited States/ ; R01 MH125252/MH/NIMH NIH HHSUnited States/ ; R35 CA253175/CA/NCI NIH HHSUnited States/ ; P30 AR070155/AR/NIAMS NIH HHSUnited States/ ; R01 CA194511/CA/NCI NIH HHSUnited States/ ; R01 AR071522/AR/NIAMS NIH HHSUnited States/ ; U01 HG009080/HG/NHGRI NIH HHSUnited States/ ; R01 CA223484/CA/NCI NIH HHSUnited States/ ; U01 DP005120/DP/NCCDPHP CDC HHSUnited States/ ; K25 HL121295/HL/NHLBI NIH HHSUnited States/ ; U01 HG012192/HG/NHGRI NIH HHSUnited States/ ; F31 HG011007/HG/NHGRI NIH HHSUnited States/ ; HHMI/Howard Hughes Medical InstituteUnited States/ ; R01 HG006399/HG/NHGRI NIH HHSUnited States/ ; T32 HG002536/HG/NHGRI NIH HHSUnited States/ ; R01 HG011345/HG/NHGRI NIH HHSUnited States/ ; R21 AI133337/AI/NIAID NIH HHSUnited States/
-
Volume:376
-
Issue:6589
-
Collection(s):
-
Main Document Checksum:urn:sha-512:3c6887506103336288640ad29373909cff6932677d33529a8d3b511e95f2c02f1317c917103d4b7bae133859bbd437636fce56d1437643ea011b0aa91080b639
-
Download URL:
-
File Type:
 [PDF
- 1.60 MB
]
[PDF
- 1.60 MB
]
Supporting Files

File Language:
English
ON THIS PAGE

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
CDC STACKS serves as an archival repository of CDC-published products including
scientific findings,
journal articles, guidelines, recommendations, or other public health information authored or
co-authored by CDC or funded partners.
As a repository, CDC STACKS retains documents in their original published format to ensure public access to scientific information.
As a repository, CDC STACKS retains documents in their original published format to ensure public access to scientific information.
You May Also Like
COLLECTION
CDC Public Access