Efficient error correction for next-generation sequencing of viral amplicons
Supporting Files
Public Domain

-
Jun 25 2012
-
Details
-
Alternative Title:BMC Bioinformatics
-
Personal Author:
-
Description:Background
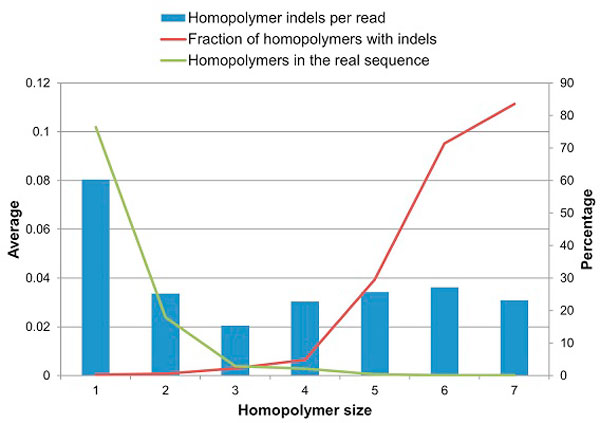
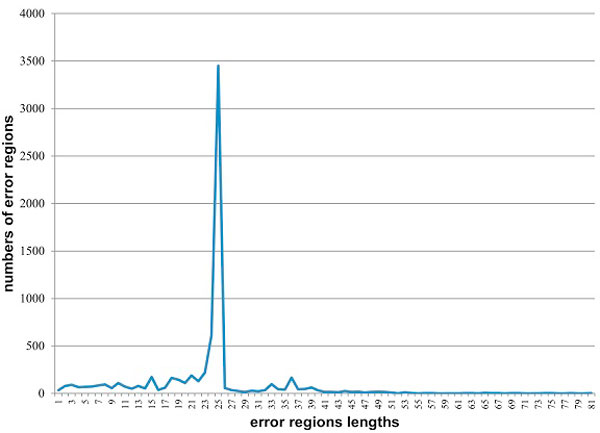
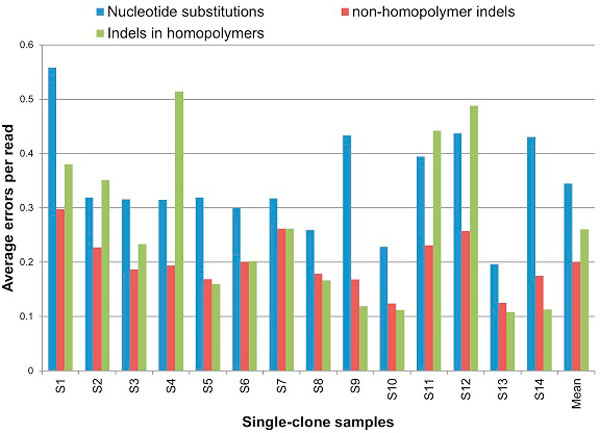
Next-generation sequencing allows the analysis of an unprecedented number of viral sequence variants from infected patients, presenting a novel opportunity for understanding virus evolution, drug resistance and immune escape. However, sequencing in bulk is error prone. Thus, the generated data require error identification and correction. Most error-correction methods to date are not optimized for amplicon analysis and assume that the error rate is randomly distributed. Recent quality assessment of amplicon sequences obtained using 454-sequencing showed that the error rate is strongly linked to the presence and size of homopolymers, position in the sequence and length of the amplicon. All these parameters are strongly sequence specific and should be incorporated into the calibration of error-correction algorithms designed for amplicon sequencing.
Results
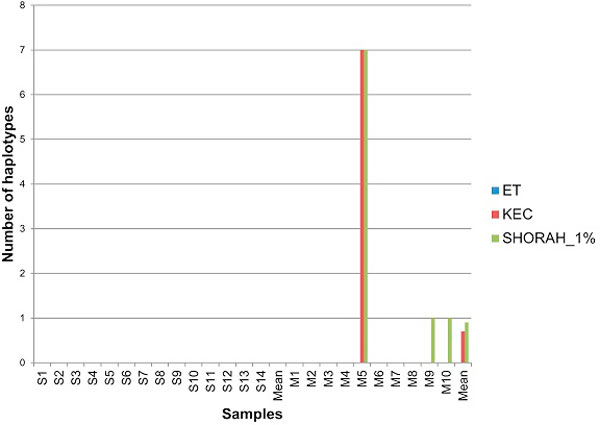
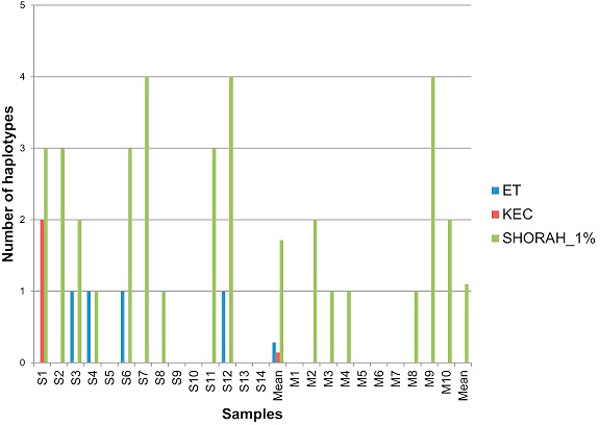
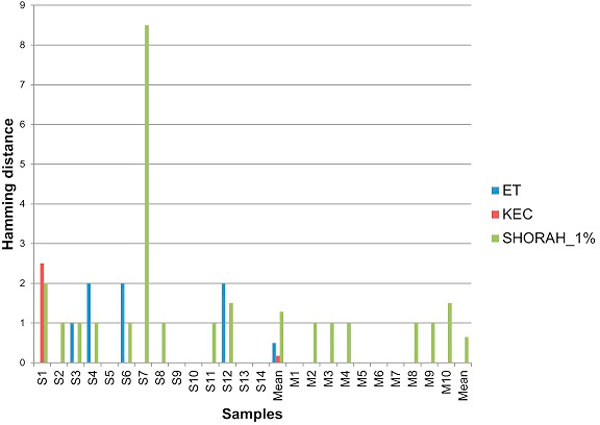
In this paper, we present two new efficient error correction algorithms optimized for viral amplicons: (i) k-mer-based error correction (KEC) and (ii) empirical frequency threshold (ET). Both were compared to a previously published clustering algorithm (SHORAH), in order to evaluate their relative performance on 24 experimental datasets obtained by 454-sequencing of amplicons with known sequences. All three algorithms show similar accuracy in finding true haplotypes. However, KEC and ET were significantly more efficient than SHORAH in removing false haplotypes and estimating the frequency of true ones.
Conclusions
Both algorithms, KEC and ET, are highly suitable for rapid recovery of error-free haplotypes obtained by 454-sequencing of amplicons from heterogeneous viruses.
-
Subjects:
-
Source:BMC Bioinformatics. 2012; 13(Suppl 10):S6.
-
Document Type:
-
Volume:13
-
Collection(s):
-
Main Document Checksum:urn:sha256:53c8dfdc09e620bf8746f8be07ea2b0e3e19630c2bef06b559478eb67ebb1ff8
-
Download URL:
-
File Type:
 [PDF
- 981.99 KB
]
[PDF
- 981.99 KB
]
Supporting Files

ON THIS PAGE

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
CDC STACKS serves as an archival repository of CDC-published products including
scientific findings,
journal articles, guidelines, recommendations, or other public health information authored or
co-authored by CDC or funded partners.
As a repository, CDC STACKS retains documents in their original published format to ensure public access to scientific information.
As a repository, CDC STACKS retains documents in their original published format to ensure public access to scientific information.
You May Also Like
COLLECTION
CDC Public Access