Human Prion Diseases in the United States
Supporting Files
Public Domain

-
Jan 01 2010
-
File Language:
English
Details
-
Alternative Title:PLoS One
-
Personal Author:
-
Description:Background
Prion diseases are a family of rare, progressive, neurodegenerative disorders that affect humans and animals. The most common form of human prion disease, Creutzfeldt-Jakob disease (CJD), occurs worldwide. Variant CJD (vCJD), a recently emerged human prion disease, is a zoonotic foodborne disorder that occurs almost exclusively in countries with outbreaks of bovine spongiform encephalopathy.
Methodology/Principal Findings
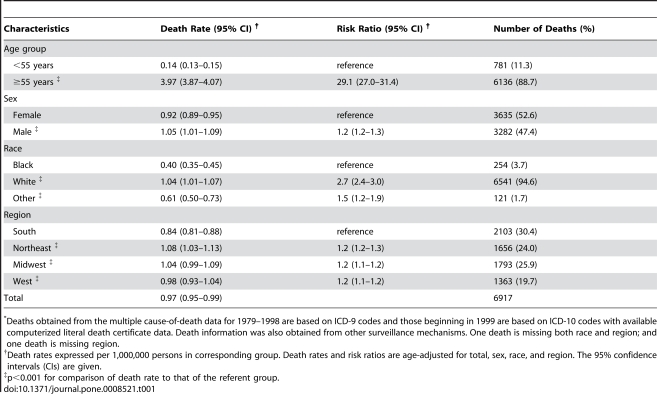
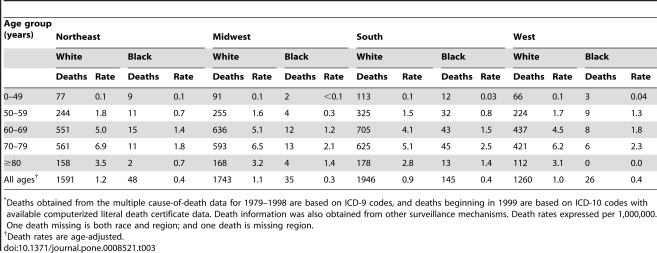
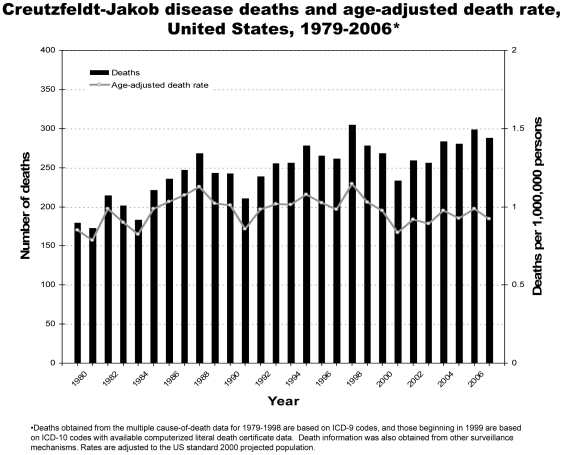
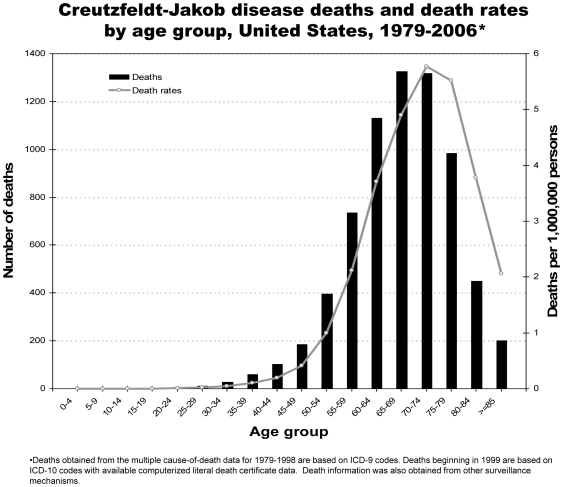
Analysis of CJD and vCJD deaths using death certificates of US residents for 1979–2006, and those identified through other surveillance mechanisms during 1996–2008. Since CJD is invariably fatal and illness duration is usually less than one year, the CJD incidence is estimated as the death rate. During 1979 through 2006, an estimated 6,917 deaths with CJD as a cause of death were reported in the United States, an annual average of approximately 247 deaths (range 172–304 deaths). The average annual age-adjusted incidence for CJD was 0.97 per 1,000,000 persons. Most (61.8%) of the CJD deaths occurred among persons ≥65 years of age for an average annual incidence of 4.8 per 1,000,000 persons in this population. Most deaths were among whites (94.6%); the age-adjusted incidence for whites was 2.7 times higher than that for blacks (1.04 and 0.40, respectively). Three patients who died since 2004 were reported with vCJD; epidemiologic evidence indicated that their infection was acquired outside of the United States.
Conclusion/Significance
Surveillance continues to show an annual CJD incidence rate of about 1 case per 1,000,000 persons and marked differences in CJD rates by age and race in the United States. Ongoing surveillance remains important for monitoring the stability of the CJD incidence rates, and detecting occurrences of vCJD and possibly other novel prion diseases in the United States.
-
Subjects:
-
Source:PLoS One. 5(1).
-
Document Type:
-
Place as Subject:
-
Volume:5
-
Issue:1
-
Collection(s):
-
Main Document Checksum:urn:sha-512:c38c883400d4e62d9021351c25968b55d8299e1094759095c97c9c65df9402c72191f71b5ae3f6e6d27f93feeabc9aec7bd3d1a64b4cacd177765a9f757bbb4f
-
Download URL:
-
File Type:
 [PDF
- 363.43 KB
]
[PDF
- 363.43 KB
]
Supporting Files

File Language:
English
ON THIS PAGE

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
CDC STACKS serves as an archival repository of CDC-published products including
scientific findings,
journal articles, guidelines, recommendations, or other public health information authored or
co-authored by CDC or funded partners.
As a repository, CDC STACKS retains documents in their original published format to ensure public access to scientific information.
As a repository, CDC STACKS retains documents in their original published format to ensure public access to scientific information.
You May Also Like
COLLECTION
CDC Public Access