Integrating Single-Molecule Experiments and Discrete Stochastic Models to Understand Heterogeneous Gene Transcription Dynamics
Supporting Files

-
Jun 12 2015
Details
-
Alternative Title:Methods
-
Personal Author:
-
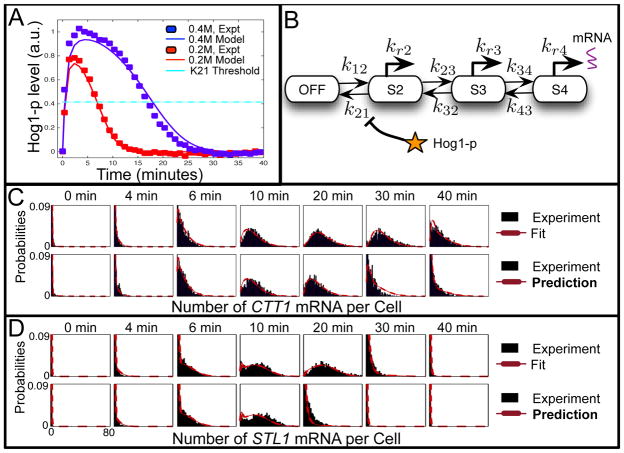
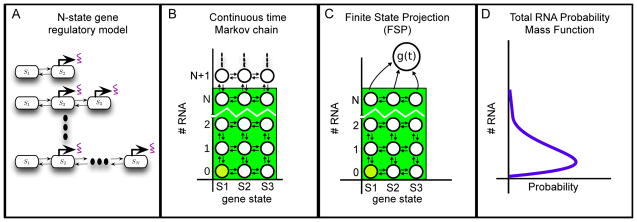
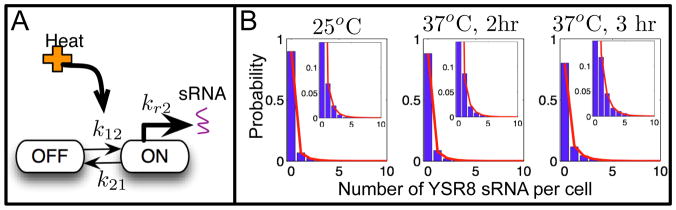
Description:The production and degradation of RNA transcripts is inherently subject to biological noise that arises from small gene copy numbers in individual cells. As a result, cellular RNA levels can exhibit large fluctuations over time and from one cell to the next. This article presents a range of precise single-molecule experimental techniques, based upon RNA fluorescence in situ hybridization, which can be used to measure the fluctuations of RNA at the single-cell level. A class of models for gene activation and deactivation is postulated in order to capture complex stochastic effects of chromatin modifications or transcription factor interactions. A computational tool, known as the finite state projection approach, is introduced to accurately and efficiently analyze these models in order to predict how probability distributions of RNA change over time in response to changing environmental conditions. These single-molecule experiments, discrete stochastic models, and computational analyses are systematically integrated to identify models of gene regulation dynamics. To illustrate the power and generality of our integrated experimental and computational approach, we explore cases that include different models for three different RNA types (sRNA, mRNA and nascent RNA), three different experimental techniques and three different biological species (bacteria, yeast and human cells).
-
Subjects:
-
Source:Methods. 85:12-21.
-
Pubmed ID:26079925
-
Pubmed Central ID:PMC4537808
-
Document Type:
-
Funding:
-
Volume:85
-
Collection(s):
-
Main Document Checksum:urn:sha256:9a88373c3ad24a3e54657eade3f152d753c7a5bb8fd2a0227bb9abe6eab94806
-
Download URL:
-
File Type:
 [PDF
- 992.29 KB
]
[PDF
- 992.29 KB
]
Supporting Files

ON THIS PAGE

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
CDC STACKS serves as an archival repository of CDC-published products including
scientific findings,
journal articles, guidelines, recommendations, or other public health information authored or
co-authored by CDC or funded partners.
As a repository, CDC STACKS retains documents in their original published format to ensure public access to scientific information.
As a repository, CDC STACKS retains documents in their original published format to ensure public access to scientific information.
You May Also Like
COLLECTION
CDC Public Access