Estimating epidemiologic dynamics from cross-sectional viral load distributions
Supporting Files

-
7 16 2021
-
File Language:
English
Details
-
Alternative Title:Science
-
Personal Author:
-
Description:INTRODUCTION:
Current approaches to epidemic monitoring rely on case counts, test positivity rates, and reported deaths or hospitalizations. These metrics, however, provide a limited and often biased picture as a result of testing constraints, unrepresentative sampling, and reporting delays. Random cross-sectional virologic surveys can overcome some of these biases by providing snapshots of infection prevalence but currently offer little information on the epidemic trajectory without sampling across multiple time points.
RATIONALE:
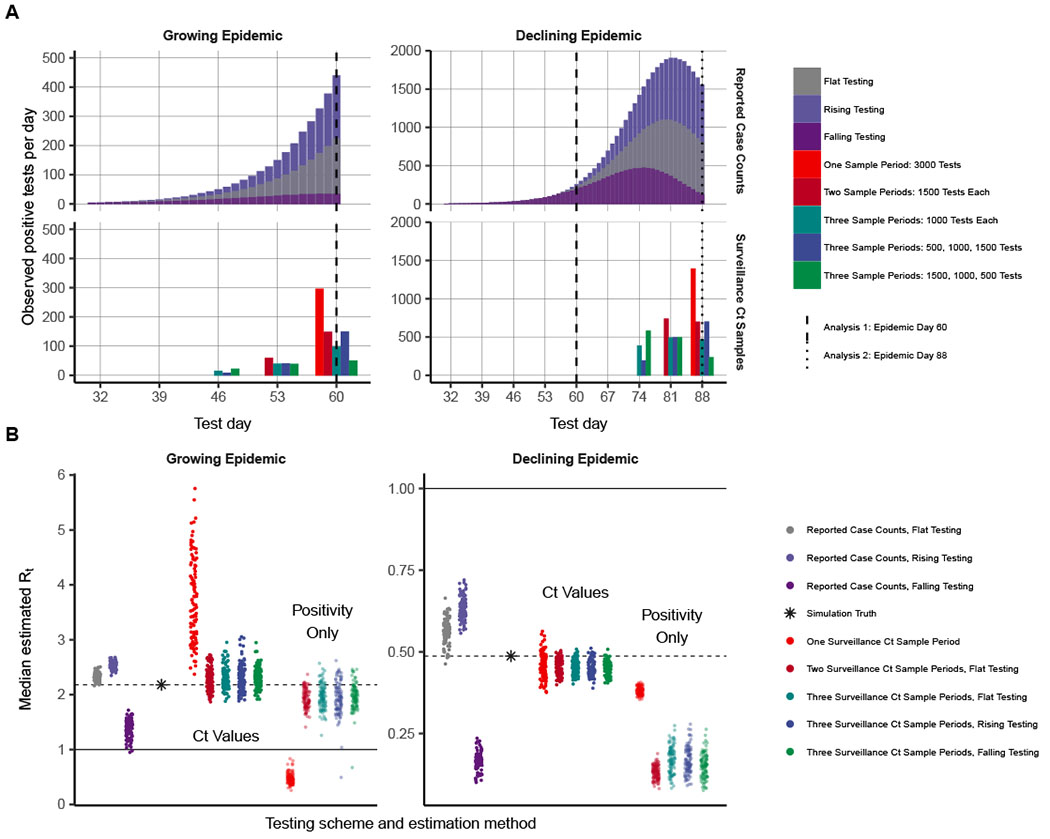
We develop a new method that uses information inherent in cycle threshold (Ct) values from reverse transcription quantitative polymerase chain reaction (RT-qPCR) tests to robustly estimate the epidemic trajectory from multiple or even a single cross section of positive samples. Ct values are related to viral loads, which depend on the time since infection; Ct values are generally lower when the time between infection and sample collection is short. Despite variation across individuals, samples, and testing platforms, Ct values provide a probabilistic measure of time since infection. We find that the distribution of Ct values across positive specimens at a single time point reflects the epidemic trajectory: A growing epidemic will necessarily have a high proportion of recently infected individuals with high viral loads, whereas a declining epidemic will have more individuals with older infections and thus lower viral loads. Because of these changing proportions, the epidemic trajectory or growth rate should be inferable from the distribution of Ct values collected in a single cross section, and multiple successive cross sections should enable identification of the longer-term incidence curve. Moreover, understanding the relationship between sample viral loads and epidemic dynamics provides additional insights into why viral loads from surveillance testing may appear higher for emerging viruses or variants and lower for out-breaks that are slowing, even absent changes in individual-level viral kinetics.
RESULTS:
Using a mathematical model for population-level viral load distributions calibrated to known features of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) viral load kinetics, we show that the median and skewness of Ct values in a random sample change over the course of an epidemic. By formalizing this relationship, we demonstrate that Ct values from a single random cross section of virologic testing can estimate the time-varying reproductive number of the virus in a population, which we validate using data collected from comprehensive SARS-CoV-2 testing in long-term care facilities. Using a more flexible approach to modeling infection incidence, we also develop a method that can reliably estimate the epidemic trajectory in even more-complex populations, where interventions may be implemented and relaxed over time. This method performed well in estimating the epidemic trajectory in the state of Massachusetts using routine hospital admissions RT-qPCR testing data—accurately replicating estimates from other sources for the entire state.
CONCLUSION:
This work provides a new method for estimating the epidemic growth rate and a framework for robust epidemic monitoring using RT-qPCR Ct values that are often simply discarded. By deploying single or repeated (but small) random surveillance samples and making the best use of the semiquantitative testing data, we can estimate epidemic trajectories in real time and avoid biases arising from nonrandom samples or changes in testing practices over time. Understanding the relationship between population-level viral loads and the state of an epidemic reveals important implications and opportunities for interpreting virologic surveillance data. It also highlights the need for such surveillance, as these results show how to use it most informatively.
-
Subjects:
-
Source:Science. 373(6552)
-
Pubmed ID:34083451
-
Pubmed Central ID:PMC8527857
-
Document Type:
-
Funding:
-
Volume:373
-
Issue:6552
-
Collection(s):
-
Main Document Checksum:urn:sha256:55e4c904b299ee884bcb4269813b203273c629fead53e98f561ce3c6f0a5e69a
-
Download URL:
-
File Type:
 [PDF
- 1.02 MB
]
[PDF
- 1.02 MB
]
Supporting Files

File Language:
English
ON THIS PAGE

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
CDC STACKS serves as an archival repository of CDC-published products including
scientific findings,
journal articles, guidelines, recommendations, or other public health information authored or
co-authored by CDC or funded partners.
As a repository, CDC STACKS retains documents in their original published format to ensure public access to scientific information.
As a repository, CDC STACKS retains documents in their original published format to ensure public access to scientific information.
You May Also Like
COLLECTION
CDC Public Access